Statins are a group of drugs taken by more than 20 million Americans each day to lower plasma low-density lipoprotein (LDL) and reduce the incidence of atherosclerotic cardiovascular disease (ACVD) and heart attacks. Statins inhibit endoplasmic reticulum (ER)-localized 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase, an enzyme that catalyzes reduction of HMG CoA to mevalonate. This reaction constitutes the rate-limiting step in synthesis of cholesterol and essential nonsterol isoprenoids including farnesyl pyrophosphate (Fpp), geranylgeranyl pyrophosphate (GGpp), ubiquinone, vitamin K2, heme, and dolichol (Fig. 1). Statin-mediated inhibition of reductase reduces cholesterol synthesis in the liver, which enhances expression of hepatic LDL-receptors that remove LDL from circulation. Although statins lower LDL by up to 50%, further reduction is limited because livers of animals and humans respond to reductase inhibition by raising the amount of the enzyme. The statin-induced increase in reductase results from reduced synthesis of sterol and nonsterol isoprenoids that inhibit the enzyme through transcriptional and post-transcriptional mechanisms. At the transcriptional level, the reductase mRNA increases owing to proteolytic activation of sterol regulatory element-binding proteins (SREBPs). At the post-transcriptional level, the increase in reductase protein is attributable to a marked decrease in its degradation rate.
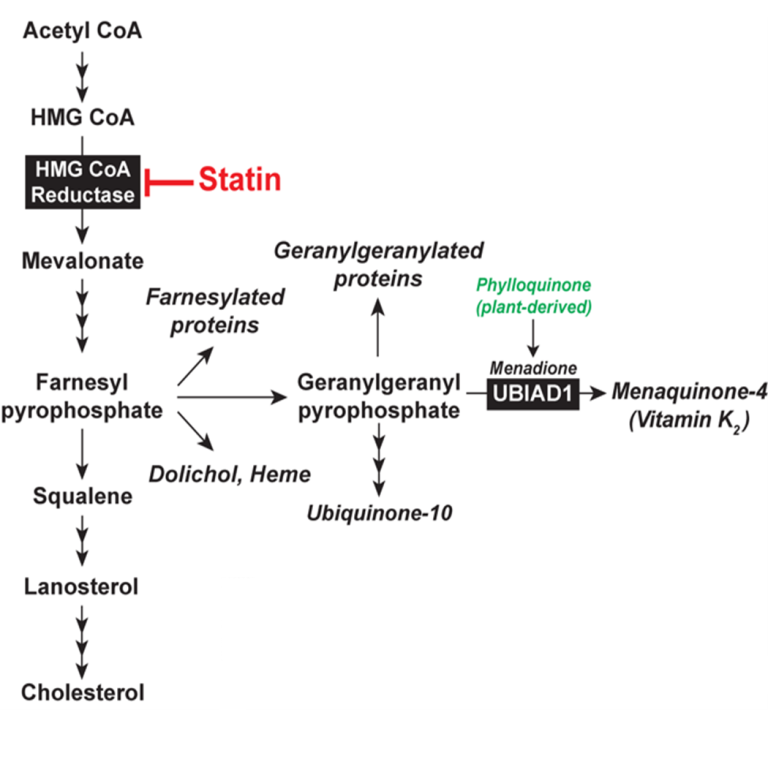 Fig. 1. The Mevalonate Pathway in Animal Cells
Fig. 1. The Mevalonate Pathway in Animal Cells Reductase is integrated in ER membranes through an N-terminal domain with 8 transmembrane helices that precedes a cytosolic catalytic domain. When sterol levels rise in those membranes, reductase is ubiquitinated and becomes degraded through a process called ER-associated degradation (ERAD). As a result, reductase molecules live for about 30 minutes. When membrane sterols are depleted by statins, reductase is no longer ubiquitinated and its half-life increases 20-fold. Our group discovered that trigger binding of the membrane domain of reductase to the ER membrane proteins Insig-1 and Insig-2. Insig-associated E3 ubiquitin ligases facilitate ubiquitination of reductase, marking it for extraction across ER membrane and release into cytosol for proteasome-mediated ERAD. Maximal ERAD requires the addition to cells of GGpp, which augments ERAD of ubiquitinated reductase by enhancing its membrane extraction. Recently, we discovered that sterols also cause a subset of reductase molecules to bind UbiA prenyltransferase domain-containing protein-1 (UBIAD1), an integral membrane prenyltransferase that utilizes GGpp to synthesize menaquinone-4 (MK-4, vitamin K2) (Fig. 1). This binding protects reductase from accelerated ERAD, permitting continued synthesis of nonsterol isoprenoids in sterol-replete cells. GGpp triggers release of UBIAD1 from reductase, allowing for maximal ERAD and translocation of UBIAD1 from the ER to the Golgi (Fig. 2). Mutations in the UBIAD1 gene cause Schnyder corneal dystrophy (SCD), an autosomal dominant eye disease characterized by corneal accumulation of cholesterol. Missense mutations altering 21 amino acids in the UBIAD1 protein have been identified.
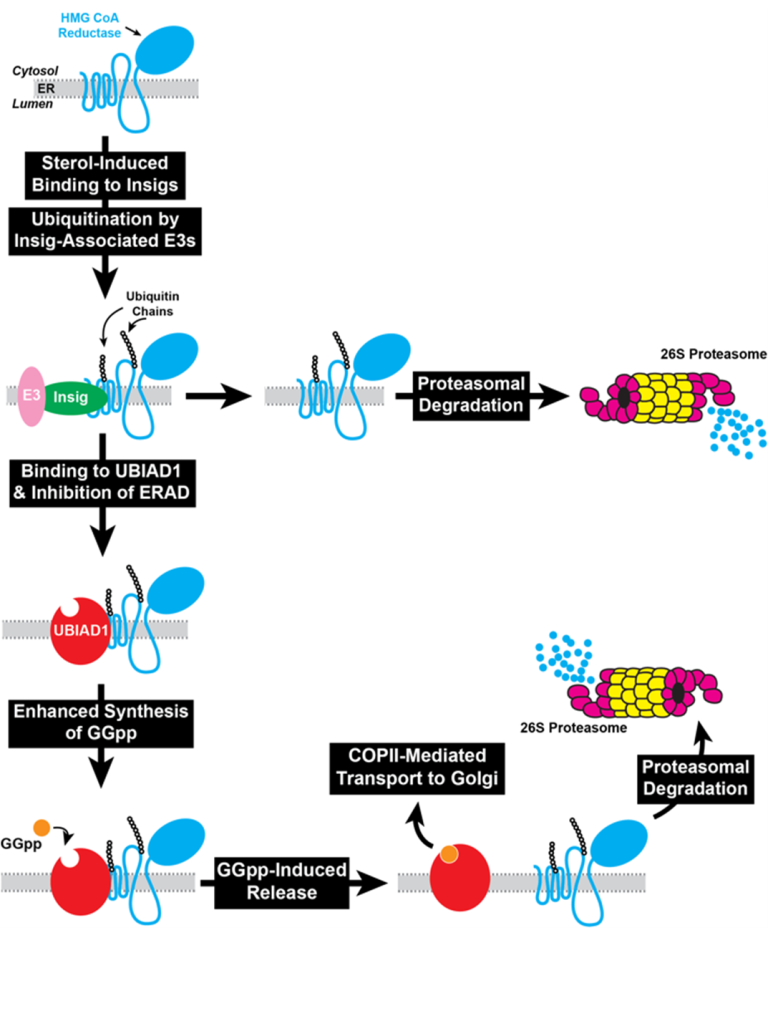 Fig. 2. UBIAD1 coordinates synthesis of sterol and nonsterol isoprenoids by inhibiting ERAD of HMG CoA reductase.
Fig. 2. UBIAD1 coordinates synthesis of sterol and nonsterol isoprenoids by inhibiting ERAD of HMG CoA reductase. SCD-associated UBIAD1 resists GGpp-induced release from reductase and remains sequestered in the ER to inhibit ERAD. The resultant inhibition of reductase ERAD contributes to accumulation of cholesterol that characterizes SCD.
The overall goals of the DeBose-Boyd Research Program are to elucidate, in molecular detail, mechanisms through which reductase senses levels of membrane-embedded sterols, how UBIAD1-mediated sensing of GGpp modulates reductase ERAD and determine the significance of the reaction in regulating metabolism of cholesterol and MK-4 in whole animals.