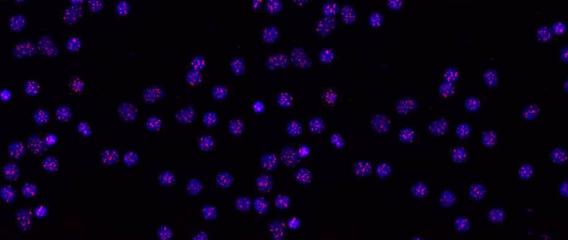
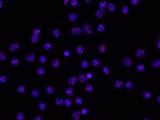
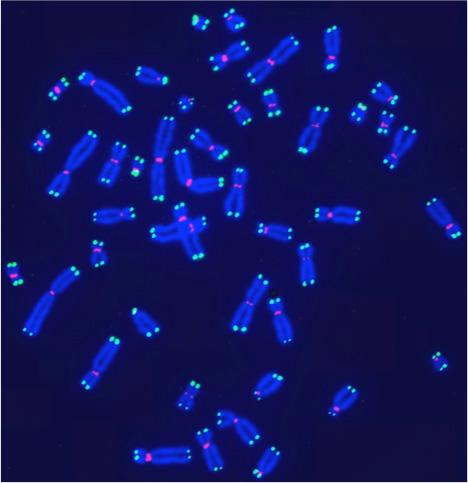
 Human telomeres on metaphase chromosomes using digital fluorescence microscopy. Metaphase chromosome spreads were prepared for quantitative fluorescence in situ hybridization (Q-FISH) analysis using oligonucleotide N3’→P5’phosphoramidate probes specific for telomere DNA sequences (green color), centromeric DNA sequences (red color) probe, and the general DNA dye DAPI (blue color).
Human telomeres on metaphase chromosomes using digital fluorescence microscopy. Metaphase chromosome spreads were prepared for quantitative fluorescence in situ hybridization (Q-FISH) analysis using oligonucleotide N3’→P5’phosphoramidate probes specific for telomere DNA sequences (green color), centromeric DNA sequences (red color) probe, and the general DNA dye DAPI (blue color). Shay Lab is interested in the relationships between aging and cancer and has focused on the role of the telomeres and telomerase in these processes. A major contribution has been the "bench to bedside" development of telomerase inhibitors that are now in advanced cancer clinical trials. We are currently testing a novel telomerase dependent telomere uncapping small molecule (Cancer Discovery, 2015), identifying methods to alter TERT (telomerase) splicing in cancer cells (Nature Communications, 2014; Cell Reports, 2013; Trends Genetics, 2014), and investigating human diseases of telomere dysfunction (called telomeropathies) which are an emerging genetic spectrum disorder (J. Cell Biol, 2014, Nucleic Acids Res, 2007, PNAS, 2007). In addition, we are moving a novel targeted therapeutic approach for colon cancer into the clinic and are involved in human cell and tissue engineering projects.
Facts About Telomeres and Telomerase
To better understand telomeres and telomerase, let's first review some basic principles of biology and genetics. The human body is an organism formed by adding many organ systems together. Those organ systems are made of individual organs. Each organ contains tissues designed for specific functions like absorption and secretion. Tissues are made of cells that have joined together to perform those special functions. Each cell is then made of smaller components called organelles, one of which is called the nucleus.
The nucleus contains structures called chromosomes that are actually "packages" of all the genetic information that is passed from parents to their children. The genetic information, or "genes," is really just a series of bases called Adenine (A), Guanine (G), Cytosine (C), and Thymine (T). These base pairs make up our cellular alphabet and create the sequences, or instructions needed to form our bodies. In order to grow and age, our bodies must duplicate their cells. This process is called mitosis. Mitosis is a process that allows one "parent" cell to divide into two new "daughter" cells. During mitosis, cells make copies of their genetic material. Half of the genetic material goes to each new daughter cell. To make sure that information is successfully passed from one generation to the next, each chromosome has a special protective cap called a telomere located at the end of its "arms.” Telomeres are controlled by the presence of the enzyme telomerase.
A telomere is a repeating DNA sequence (for example, TTAGGG) at the end of the body's chromosomes. The telomere can reach a length of 15,000 base pairs. Telomeres function by preventing chromosomes from losing base pair sequences at their ends. They also stop chromosomes from fusing to each other. However, each time a cell divides, some of the telomere is lost (usually 25-200 base pairs per division). When the telomere becomes too short, the chromosome reaches a "critical length" and can no longer replicate. This means that a cell becomes "old" and dies by a process called apoptosis. Telomere activity is controlled by two mechanisms: erosion and addition. Erosion, as mentioned, occurs each time a cell divides. Addition is determined by the activity of telomerase.
A telomere is a repeating DNA sequence (for example, TTAGGG) at the end of the body's chromosomes. The telomere can reach a length of 15,000 base pairs. Telomeres function by preventing chromosomes from losing base pair sequences at their ends. They also stop chromosomes from fusing to each other. However, each time a cell divides, some of the telomere is lost (usually 25-200 base pairs per division). When the telomere becomes too short, the chromosome reaches a "critical length" and can no longer replicate. This means that a cell becomes "old" and dies by a process called apoptosis. Telomere activity is controlled by two mechanisms: erosion and addition. Erosion, as mentioned, occurs each time a cell divides. Addition is determined by the activity of telomerase.
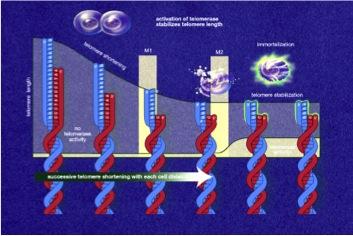 Telomerase, also called telomere terminal transferase, is an enzyme made of protein and RNA subunits that elongates chromosomes by adding TTAGGG sequences to the end of existing chromosomes. Telomerase is found in fetal tissues, adult germ cells, and also tumor cells. Telomerase activity is regulated during development and has a very low, almost undetectable activity in somatic (body) cells. Because these somatic cells do not regularly use telomerase, they age. The result of aging cells is an aging body. If telomerase is activated in a cell, the cell will continue to grow and divide. This "immortal cell" theory is important in two areas of research: aging and cancer.
Telomerase, also called telomere terminal transferase, is an enzyme made of protein and RNA subunits that elongates chromosomes by adding TTAGGG sequences to the end of existing chromosomes. Telomerase is found in fetal tissues, adult germ cells, and also tumor cells. Telomerase activity is regulated during development and has a very low, almost undetectable activity in somatic (body) cells. Because these somatic cells do not regularly use telomerase, they age. The result of aging cells is an aging body. If telomerase is activated in a cell, the cell will continue to grow and divide. This "immortal cell" theory is important in two areas of research: aging and cancer. Cellular aging, or senescence, is the process by which a cell becomes old and dies. It is due to the shortening of chromosomal telomeres to the point that the chromosome reaches a critical length. Cellular aging is analogous to a wind up clock. If the clock stays wound, a cell becomes immortal and constantly produces new cells. If the clock winds down, the cell stops producing new cells and dies. Our cells are constantly aging. Being able to make the body's cells live forever certainly creates some exciting possibilities. Telomerase research could therefore yield important discoveries related to the aging process.
Cancer cells are a type of malignant cell. The malignant cells multiply until they form a tumor that grows uncontrollably. Telomerase has been detected in human cancer cells and is found to be 10-20 times more active than in normal body cells. This provides a selective growth advantage to many types of tumors. If telomerase activity was to be turned off, then telomeres in cancer cells would shorten, just like they do in normal body cells. This would prevent the cancer cells from dividing uncontrollably in their early stages of development. In the event that a tumor has already thoroughly developed, it may be removed and anti-telomerase therapy could be administered to prevent relapse. In essence, preventing telomerase from performing its function would change cancer cells from "immortal" to "mortal."
Knowing what we have just learned about telomeres and telomerase, it can be said that scientists are on the verge of discovering many of telomerase's secrets. In the future, their research in the area of telomerase could uncover valuable information to combat aging, fight cancer, and even improve the quality of medical treatment in other areas such as skin grafts for burn victims, bone marrow transplants, and heart disease. Who knows how far this could go?
Telomere overhang generation processes and the end-replication problem
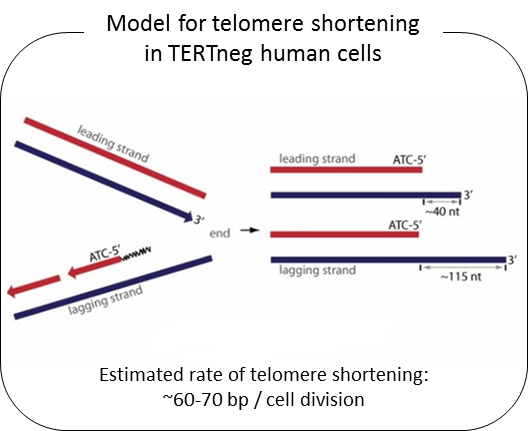
Telomeres are composed of TTAGGG repeated sequences located at the end of chromosomes. In human somatic cells, cell divisions are accompanied with progressive telomere length shortening due to lack of or insufficient telomerase activity (termed the end-replication problem). It is generally believed that telomere length shortening results from the removal of the RNA primer binding site at the Okazaki fragment located in lagging DNA strand synthesis. If that were correct, the rate of telomere shortening would only be around 8-10 bp per cell division (RNA primer size ÷ 2 = (16 ~ 20 nt) ÷ 2). Nevertheless, telomerase negative human cells show telomere shortening rates of ~60-70 bp per cell division. From previous studies, we found that telomere overhang generation processes may be important in determining telomere shortening rates. We found that the telomere lagging strand has an overhang of ~110 nt and the leading strand has an overhang of ~30 nt. These results can explain the telomere shortening rate in telomerase negative cells [(lagging strand overhang 110 nt + leading strand overhang 30 nt)÷2 = ~70nt].

We have recently found that human telomere overhangs in telomerase-negative cells are processed by multiple steps and leading/lagging strands are produced by distinct mechanisms. Leading strand overhangs have an early process that occurs throughout S phase, and a late process that occurs in late S/G2 phase. In contrast, lagging strand overhang processes are completed as soon as the end is replicated. At present, we are determining the molecular factors involved in overhang generation processes using biochemical approaches as well as a new system for introducing telomerase negative conditions in various human cell lines using advanced techniques of genome editing.
Telomerase Regulation
Telomerase activity is tightly controlled in humans because telomere length imposes a limit on the number of times that any given cell can divide. Almost all cancer cells have reactivated telomerase in order to overcome this replicative limit via poorly understood mechanisms. One aim is to elucidate the regulatory mechanisms that confine telomerase expression to a subset of stem cells and germ cells and how those regulatory mechanisms are circumvented by cancer cells. Telomere length and telomerase expression also play a role in aging (telomeres become progressively shorter over time), and so a greater understanding of the telomerase regulatory network may also improve our understanding of the molecular mechanisms of aging and lead to potential anti-aging therapies.
Telomerase regulation in normal cells
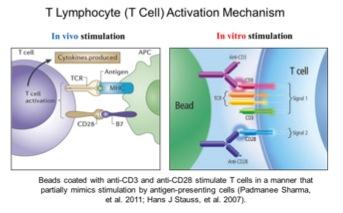
Telomerase activity is usually undetectable in most human tissues, except for some proliferating stem-like cells in rapid turnover tissues. Previous reports have shown that mitogen stimulated human T lymphocytes transiently turn on telomerase activity that may reduce the rate of telomere loss during rapid proliferation. However, telomerase activation is transient in T-cells as opposed to cancer cells, and is only maintained for a few days even with continual mitogen stimulation. After approximately 4 days of stimulation, telomerase activity greatly decreases and eventually T-cells stop proliferating. With increased human age, T-lymphocytes show progressive telomere shortening. Almost all cancer cells activate telomerase, it does not turn off, cells achieve unlimited proliferation and telomeres do not further shorten. Currently it is unclear how telomerase activation is regulated in normal cells (such as in T lymphocytes) and how this regulation is hijacked by cancer cells.
Telomerase alternative splicing in normal human lymphocytes
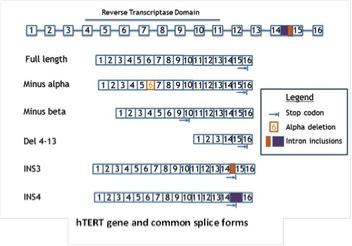
Our lab has demonstrated that hTERT alternative splicing is a potential mechanism for telomerase regulation. In our recent studies, we found that changes of telomerase activity after T lymphocyte stimulation corresponded with the expression shifts of several hTERT splicing variants. By 72 hours, the ratio of catalytically active telomerase was increased compared to the non-functional splice variants (minus alpha and minus beta). This observation further emphasizes the potential role of hTERT alternative splicing in telomerase regulation in normal T lymphocytes. Taking advantage of the T lymphocyte stimulation model, we aim to study telomerase regulatory mechanisms in normal cells. The elucidation of how telomerase is regulated reversibly in primary proliferating transiently amplifying cells may facilitate our understanding of the potential mechanism(s) that cancer cells use to maintain telomerase activation. As part of these studies we will be examining T-cells from various aged individuals including super centenarians.
Telomerase alternative splicing in normal and cancer cells
Telomerase is regulated on many levels—from traditional transcriptional control to epigenetic regulation. Expression of hTERT is also regulated at the level of alternative splicing—with the full-length mRNAs being a minor component. We are determining the identities of all hTERT splice variants, the context in which they are expressed, and their function in normal and cancer cells. Since most splice forms are catalytically inactive and some are even potentially inhibitory, small molecules to increase the fraction of inactive hTERT splicing may be a viable option to produce shortening of telomeres in cancer cells, while increasing the fraction of active hTERT splicing could be used to increase the regenerative potential of stem cells.
Telomerase Inhibitors
One of the hallmarks of cancer is the limitless proliferation of cancer cells, which is related to telomerase reactivation. Since most cancer cells express telomerase to bypass the replicative limit imposed by telomere length, development and testing of new compounds that target telomerase expressing cancer cells, not normal cells, is a high-priority avenue for emerging cancer therapies. Our lab works on different approaches for telomerase targeted therapy (e.g. direct telomerase inhibition and telomerase mediated telomere targeting approaches). We evaluate their clinical utility in tissue culture, mouse xenografts, and in clinical trials with both industrial and academic partners.
Telomere Position Effects
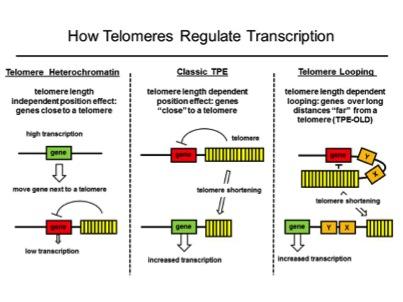
We found that genes at long distances (up to 10Mb) from telomeres may be regulated differently than the classic telomere position effect (TPE) mechanism. Classic TPE is produced by telomeric heterochromatin invading the subtelomeric region and silencing nearby genes. We discovered that interferon stimulating gene 15 (ISG15, over 1 MB from the telomere) is regulated by telomere length but genes closer to the telomere were not silenced by classic TPE. We called this phenomenon telomere position effect over long distances (or TPE-OLD) to distinguish it from classic TPE. The changes in genomic structure at various loci might provide novel insights into how the regulation of genes in somatic cells is changed during telomere shortening as part of normal aging and how this might lead to a permissive environment for tumor development.
Lung Cancer Therapeutics
National Aeronautical Space Administration (NASA) radiation research program is focused on enabling long duration exploration of space within acceptable risk limits. Astronauts will be exposed to radiation doses that may result in adverse long-term health risks such as damage to the nervous and cardiovascular systems and an increased risk of cancer development. During space exploration, astronauts are exposed to varying types of radiation including Galactic Cosmic Radiation (GCR) and High Atomic Number and Energy (HZE) particles. Space radiation differs from terrestrial radiation in several aspects such as ionizing pattern and energy deposition. These differences do not allow the extrapolation of our understanding of risks and mechanisms from terrestrial radiation exposure to that in space. Because there are no human epidemiological data for these radiation types, risk estimation must be generated from mechanistic details based on radiation physics and on molecular, cellular, tissue and organismal radiation biology related to exposure.
Biologically significant data collected and compared to radiation type, dose, dose-rates, and energies provide valuable information to more effectively assess acceptable risk limits for astronauts on long-term space missions. Utilizing lung cancer mouse models (K-ras and EGFR), colon cancer susceptible mouse models (CPC;APC) and system biology approaches, our research is focused on obtaining relevant biological information on radiation-induced effects such as DNA damage/processing, signal transduction, cell cycle controls, cellular differentiation, and chronic inflammation. Our research is leading to quantitative assessments of radiation effects and a more comprehensive understanding of mechanism(s) involved in space radiation associated health risk so we can develop biological countermeasures to reduce these risks.
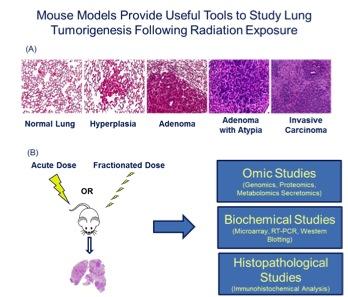 Figure 2. (A) The KrasLA1 mouse model of lung tumorigenesis recapitulates many aspects of human lung cancer development and provides an invaluable tool to study disease etiology. (B) Utilizing lung cancer susceptibility models and system biology approaches, our research is focused on obtaining relevant biological information on radiation-induced effects.
Figure 2. (A) The KrasLA1 mouse model of lung tumorigenesis recapitulates many aspects of human lung cancer development and provides an invaluable tool to study disease etiology. (B) Utilizing lung cancer susceptibility models and system biology approaches, our research is focused on obtaining relevant biological information on radiation-induced effects. Tissue Engineering
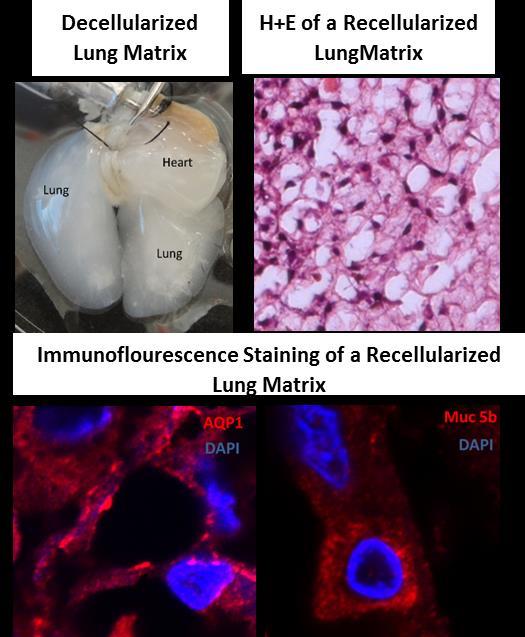
The Shay lab tissue engineering group seeds life-extended or immortalized human cells into a variety of three-dimensional (3-D) substrates in order to promote differentiation. The long-term goal is not only to produce functional organs (lung, heart, kidney) in vitro, but also to be able to have the cells express molecules that produce immune tolerance so that tissue-matching would no longer be required. Our most advanced model system uses bronchial lung epithelial cells seeded onto a decellularized lung matrix. This project has two broad aims: 1) develop novel cell reagents for lung tissue engineering and lung disease (cancer, lung fibrosis) modeling applications; 2) create 3-D tissue constructs using mutant adult lung progenitor cells for disease modeling (such as cystic fibrosis, pulmonary fibrosis and cancer) and normal progenitor cells for regenerative medicine applications.
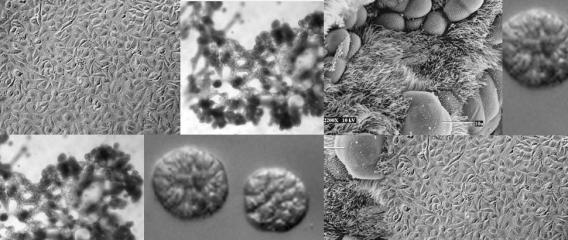
Primary cells isolated from adult patient lungs can normally only be passaged a few times before losing the ability to differentiate, making it difficult to isolate a sufficient number of cells for disease studies or tissue engineering. To address this, we have developed normal human bronchial epithelial cells (HBECs) which have been either immortalized by ectopic expression of CDK4 and hTERT/telomerase (HBEC3KTs) or conditionally life-extended using Rho-Kinase inhibitors while grown on an irradiated fibroblast feeder layer (or conditioned medium from irradiated feeder layers). These immortalized or conditionally life-extended cells are multipotent and form different lineages based on cues from the extracellular microenvironment. HBEC3KTs can form a well differentiated epithelium composed of basal, goblet, and ciliated cells when placed at the air-liquid interface (Figure 1A, B). HBEC3KTs can also form bronchial organoids/cyst-like structures when grown inside Matrigel® or highly branched structures when seeded on top of Matrigel® with a lung fibroblast feeder layer below the matrix (Figure 1C, D). In addition to primary bronchial cells from normal lung donors, we have isolated bronchial cells from patients with cystic fibrosis and lung cancer. We have thus created cellular reagents for the study of normal and diseased lung development that can be maintained in culture for a significantly extended period of time compared to traditional studies with primary bronchial cells.
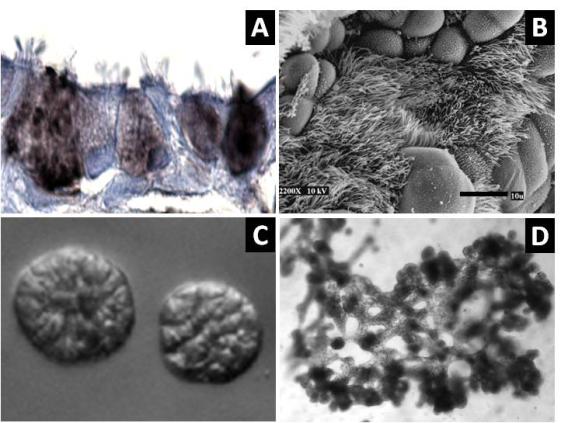 Figure 1. Differentiation of HBEC3KTs in different extracellular conditions. (A) Paraffin cross-section of well differentiated air-liquid interface (ALI) culture with immuno-histochemistry stain for MUC5AC+ goblet cells interspersed between ciliated cells. (B) Scanning electron micrograph also shows well differentiated goblet and ciliated cells at ALI. Scale = 10 µm (C) Phase contrast picture of 5 day bronchial organoids 50 µm in diameter embedded in Matrigel® with a fibroblast feeder layer. (D) Phase contrast picture of 10 day highly branched organotypic structure on top of Matrigel® with a fibroblast feeder layer below.
Figure 1. Differentiation of HBEC3KTs in different extracellular conditions. (A) Paraffin cross-section of well differentiated air-liquid interface (ALI) culture with immuno-histochemistry stain for MUC5AC+ goblet cells interspersed between ciliated cells. (B) Scanning electron micrograph also shows well differentiated goblet and ciliated cells at ALI. Scale = 10 µm (C) Phase contrast picture of 5 day bronchial organoids 50 µm in diameter embedded in Matrigel® with a fibroblast feeder layer. (D) Phase contrast picture of 10 day highly branched organotypic structure on top of Matrigel® with a fibroblast feeder layer below. The decellularized matrix of complex tissues preserves the tensional, extracellular molecular, and ultrastructure cues of the original organ. Cells seeded onto these matrices rapidly differentiate and form functional tissues. One current model decellularizes a mouse lung by perfusion with detergent, seeds life-extended bronchial basal cells into the tracheal cavity, and cultures the seeded lung in a pressurized bioreactor with perfusion of media through the vasculature. We are using this system to model normal and diseased lung development in an ex-vivo environment and to pursue tissue engineering of functional lungs for regenerative medicine applications.
Colon Cancer Therapeutics
Mutations in the gene encoding the Adenomatous Polyposis Coli (APC) protein are a highly frequent and early event in colorectal cancers (CRC). Over 90 percent of mutations in the APC gene generate stable truncated gene products, leading to activation of the Wnt signaling pathway and deregulation of multiple cellular processes. However, there have been no therapeutic approaches directly targeting these APC mutations. We have performed a chemical screen employing a series of isogenic normal and partially transformed immortalized human colonic epithelial cells (HCECs) to identify small molecules that are selectively toxic to CRC lines with APC truncations while sparing cells with wild type (WT) APC. The lead compounds and their analogs exhibit antitumor activity in both xenograft and genetically engineered colorectal cancer mouse models. Current work is focusing on the investigation of the mechanism of action of these compounds and optimization of the lead compounds for future translational development as drugs for clinical testing.
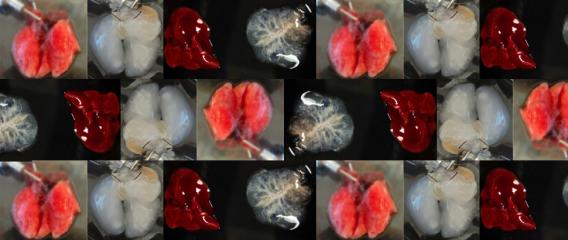
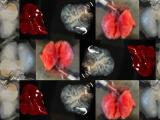
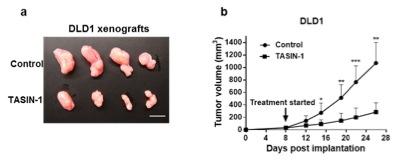 Figure 1. - a. Tumor sizes of TASIN-1 treated DLD1 xenografts (below) are smaller than those of control mice (above). Scale bar, 10mm. b. TASIN-1 significantly reduces tumor growth rate of DLD1 xenografts.
Figure 1. - a. Tumor sizes of TASIN-1 treated DLD1 xenografts (below) are smaller than those of control mice (above). Scale bar, 10mm. b. TASIN-1 significantly reduces tumor growth rate of DLD1 xenografts.