Research
Molecular mechanism of ferroptosis in development of human diseases
Our cells need both iron and polyunsaturated fatty acids for their survival. This requirement, however, creates a fundamental biochemical conflict in that iron can catalyze peroxidation of polyunsaturated fatty acids through a self-amplifying chain reaction. This reaction will lead to the accumulation of phospholipids containing peroxidized fatty acyl chains, which eventually cause cell death through ferroptosis by disrupting membrane integrity (Fig. 1). Our cells are well aware of this vulnerability, and they develop multiple strategies to guard against ferroptosis (Fig. 1). However, under certain stress conditions, these defenses could fail, leading to tissue damage caused by ferroptosis. Nevertheless, owing to the lack of a biomarker to detect ferroptotic cells under physiological conditions, the exact diseases caused by ferroptosis have been difficult to identify.
We recently addressed this challenge by identifying hyperoxidized PRDX3 as a marker for ferroptosis. In healthy cells, PRDX3 is a mitochondrial peroxiredoxin using its active site cysteine to reduce peroxides including lipid peroxides. Under ferroptotic stress, the accumulation of lipid peroxides in mitochondria triggers hyperoxidation of PRDX3, a reaction converting the active site cysteine thiol to sulfinic or sulfonic acid (Fig. 2A). Importantly, hyperoxidized PRDX3 is robustly induced only by ferroptosis but not other cell death pathways or mitochondrial damage unrelated to ferroptosis, supporting the application of hyperoxidized PRDX3 as a ferroptosis marker.
The identification of this ferroptosis marker allowed us to determine that alcoholic liver disease (ALD) caused liver damage primarily through ferroptosis (Fig. 2B). ALD, one of the most prevalent chronic liver diseases, encompasses a spectrum of disorders including advanced ALD associated with high mortality without effective treatment. My lab is currently interested in understanding how overconsumption of alcohol causes ferroptosis of liver cells so novel treatments of advanced ALD could be developed by strategies that inhibit ferroptosis. Our recent development of a mouse model of advanced ALD, as well as the establishment of cell-based analysis to study cell biology and biochemistry of ferroptosis, has made it possible to pursue these investigations.
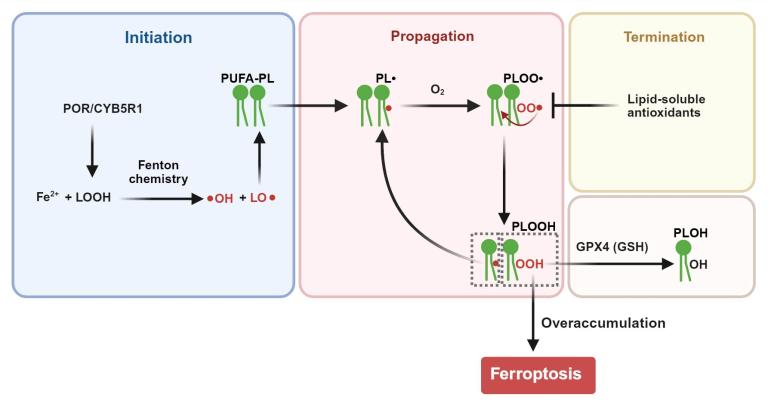
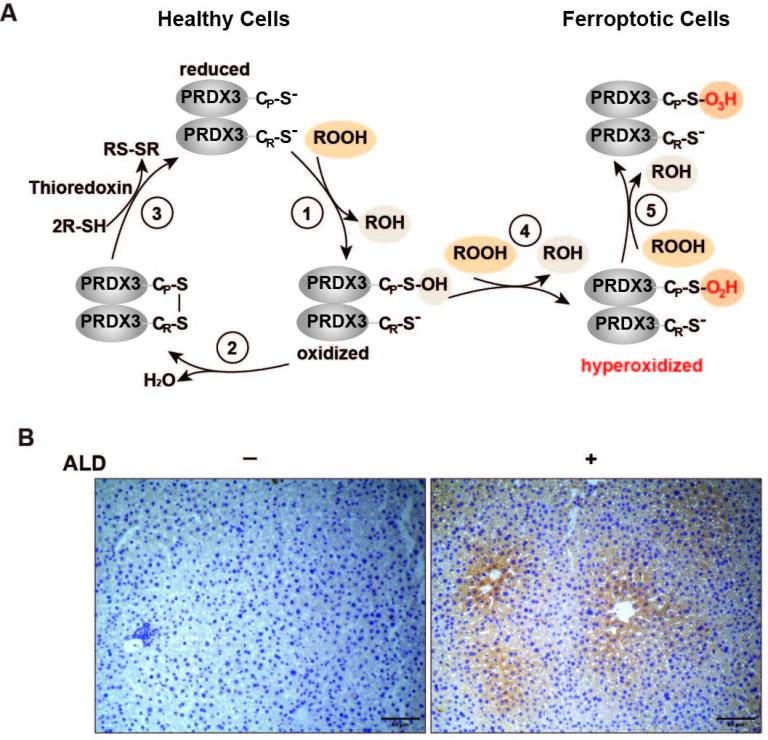
Fig. 1. Phospholipid peroxidation triggers ferroptosis
Lipid peroxidation proceeds through three stages. During initiation, Fe2+ reacts with lipid hydroperoxides (LOOH) produced by oxidoreductases such as POR and CYB5R1via Fenton chemistry to generate hydroxyl (•OH) and alkoxyl (LO•) radicals. In the propagation phase, LO• abstracts bis-allylic hydrogen atoms from PUFA-containing PLs (PUFA-PLs), generating PL radicals (PL•), which react with oxygen to form peroxyl radicals (PLOO•). The PLOO• produced attack neighboring PUFA-PLs to generate more PL•, thereby establishing a self-amplifying chain reaction that leads to accumulation of PL hydroperoxides (PLOOH). Lipid-soluble antioxidants terminate PL peroxidation by neutralizing PLOO•. GPX4, an enzyme critical for protection against ferroptosis, detoxifies PLOOH by reducing PLOOH to PL alcohols (PLOH). When the rate of lipid peroxidation exceeds this detoxification capacity, excess PLOOH compromises membrane integrity thus triggering ferroptosis.
Fig. 2. The presence of ferroptotic cells in livers with ALD through detection hyperoxidized PRDX3, the ferroptosis marker
- A. In healthy cells, PRDX3 uses a catalytic Cys thiol (Cp) to reduce peroxides. The Cys sulfenic acid (Cys-SOH) produced by the reaction leads to formation of a disulfide-linked homodimer, and the reduction of which allows the enzymes to continue their catalytic cycle (reactions 1-3). Upon accumulation of excess lipid peroxides in ferroptotic cells, the rate of disulfide bond formation is not fast enough to prevent the Cys-SOH in PRDX3 from further oxidation by the peroxides to generate Cys sulfinic (Cys-SO2H) and sulfonic acid (Cys-SO3H), the hyperoxidation products of PRDX3 (reactions 4 and 5).
- B. Detection of ferroptotic cells in mouse livers with ALD through immunohistochemistry with an antibody that recognizes hyperoxidized PRDX3.
Meet the PI & Lab Members

Jin Ye, Ph.D.
Jin Ye received a M.S. degree in Biochemistry from Case Western Reserve University in 1995. He was mentored by Nobel laureates Michael Brown and Joseph Goldstein at UT Southwestern, obtaining his Ph.D. in Cell Regulation in 2000, and continuing in a postdoctoral fellowship from 2000 to 2004.He joined the faculty at the UT Southwestern Medical Center in 2004 as an Assistant Professor. He was promoted to Professor in 2022.
Dr. Ye’s research interests are to understand mechanism of diseases caused by abnormal lipid metabolism. Currently his lab is investigating how ferroptosis, a cell death pathway caused by iron-catalyzed peroxidation of polyunsaturated fatty acids, in development of human diseases such as alcoholic liver disease. His lab has employed multiple approaches ranging from in vitro biochemical assays to mouse models of the human diseases to development novel treatments of diseases caused by ferroptosis.
Yanchao Xu
Senior Research Scientist
Kosuke Kamemura
Postdoctoral Researcher
Lori Nguyen
Research Associate
Publications
- Ferroptosis: The demise of cells through phospholipid peroxidation.
Cui, S., and Ye, J. Adv Sci 2026 e24387. - Protection against ferroptosis through maintaining homeostasis of docosahexaenoate-containing phospholipids.
Deng Y, Vale G, Liang Y, Cui S, Xu S, McDonald JG, Ye J. Mol Cell 2025 Sep 18 3474-3485 - Advanced alcoholic liver disease driven by a pro-ferroptotic diet.
Liang Y, Xu Y, Virostek M, Johnson A, Evers B, Deng Y, Meng Y, McDonald JG, Scherer PE, Cui S, Ye J. J Lipid Res 2025 66 100898 - MAFLD: A ferroptotic disease.
Cui S, Ye J. Trends Mol Med 2025 Sep S1471-4914(25)00194-7 - 5H7c: A rabbit monoclonal antibody detecting ferroptotic cells.
Cui, S., Donnelly, L., Ghai, A., Achilefu, S., and Ye, J. Mol Cell 2024 84, 4471111-4472 - 7-Dehydrocholesterol: A sterol shield against an iron sword.
Cui, S. and Ye, J. Mol Cell 2024 84 1183-1185 - Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases.
Cui S, Ghai A, Deng Y, Li S, Zhang R, Egbulefu C, Liang G, Achilefu S, Ye J, Mol Cell 2023 Oct 83 3931-3939.e3935 - Topological regulation of a transmembrane protein by luminal-to-cytosolic retrotranslocation of glycosylated sequence.
Wang J, Han S, Ye J, Cell Rep 2023 Mar 42 4 112311 - FAF1 blocks ferroptosis by inhibiting peroxidation of polyunsaturated fatty acids.
Cui S, Simmons G, Vale G, Deng Y, Kim J, Kim H, Zhang R, McDonald JG, Ye J, Proc Natl Acad Sci U S A 2022 Apr 119 17 e2107189119 - Identification of TRAMs as Sphingolipid-Binding Proteins Using a Photoactivatable and Clickable Short Chain Ceramide Analog.
Deng Y, You L, Lu Y, Han S, Wang J, Vicas N, Chen C, Ye J, J Biol Chem 2021 Nov 101415 - Regulating G protein-coupled receptors by topological inversion.
Denard B, Han S, Kim J, Ross EM, Ye J, Elife 2019 Mar 8 - Identification of residues critical for topology inversion of the transmembrane protein TM4SF20 through regulated alternative translocation.
Wang J, Kinch LN, Denard B, Lee CE, Esmaeilzadeh Gharehdaghi E, Grishin N, Ye J J. Biol. Chem. 2019 Feb - Inverting the Topology of a Transmembrane Protein by Regulating the Translocation of the First Transmembrane Helix.
Chen Q, Denard B, Lee CE, Han S, Ye JS, Ye J Mol. Cell 2016 Aug 63 4 567-78 - Unsaturated Fatty Acids Stimulate Tumor Growth through Stabilization of β-Catenin.
Kim H, Rodriguez-Navas C, Kollipara RK, Kapur P, Pedrosa I, Brugarolas J, Kittler R, Ye J Cell Rep 2015 Oct 13 3 495-503 - UAS Domain of Ubxd8 and FAF1 Polymerizes upon Interaction with Long Chain Unsaturated Fatty Acids.
Kim H, Zhang H, Meng D, Russell G, Lee JN, Ye J J. Lipid Res. 2013 May - Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1.
Denard B, Lee C, Ye J Elife 2012 1 e00090 - Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis.
Lee JN, Kim H, Yao H, Chen Y, Weng K, Ye J, Proc Natl Acad Sci U S A 2010 Dec 107 50 21424-9 - Identification of CREB3L1 as a Biomarker Predicting Doxorubicin Treatment Outcome.
Denard B, Pavia-Jimenez A, Chen W, Williams NS, Naina H, Collins R, Brugarolas J, Ye J PLoS ONE 2015 10 6 e0129233 - Sustained induction of collagen synthesis by TGF-β requires regulated intramembrane proteolysis of CREB3L1.
Chen Q, Lee CE, Denard B, Ye J PLoS ONE 2014 9 10 e108528 - Nrf1 to the rescue.
Ye J Elife 2014 3 e02062 - Cellular responses to excess fatty acids: focus on ubiquitin regulatory X domain-containing protein 8.
Kim H, Ye J Curr. Opin. Lipidol. 2013 Dec - Roles of regulated intramembrane proteolysis in virus infection and antiviral immunity.
Ye J Biochim. Biophys. Acta 2013 Dec 1828 12 2926-32 - Sufficient production of geranylgeraniol is required to maintain endotoxin tolerance in macrophages.
Kim J, Lee JN, Ye J, Hao R, Debose-Boyd R, Ye J J. Lipid Res. 2013 Dec 54 12 3430-7 - Epigenetic silencing of antiviral genes renders clones of Huh-7 cells permissive for hepatitis C virus replication.
Chen Q, Denard B, Huang H, Ye J J. Virol. 2013 Jan 87 1 659-65 - Getting to grips with hepatitis.
Chen ZJ, Ye J Elife 2012 1 e00301 - The Membrane-Bound Transcription Factor CREB3L1 Is Activated in Response to Virus Infection to Inhibit Proliferation of Virus-Infected Cells.
Denard B, Seemann J, Chen Q, Gay A, Huang H, Chen Y, Ye J Cell Host Microbe 2011 Jul 10 1 65-74 - Regulated ERAD of a polytopic protein: p97 recruits proteasomes to insig-1 before extraction from membranes.
Ikeda, Y., Demartino, G.N., Brown, M.S., Lee, J.N., Goldstein, J.L., Ye, J. J. Biol. Chem. October 2009 284: 34889-34900 - Unsaturated fatty acids inhibit proteasomal degradation of insig-1 at a post-ubiquitination step.
Lee, J.N., Zhang, X., Feramisco, J.D., Gong, Y., Ye, J. J. Biol. Chem. October 2008 283: 33772-33783 - Long chain acyl-CoA synthetase 3-mediated phosphatidylcholine synthesis is required for assembly of very low density lipoproteins in human hepatoma Huh7 cells
Yao, H. and Ye, J. J. Biol. Chem. 2008 283 849-854 - Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus.
Ye, Jin PLoS Pathog. 2007 3 1017-1022 - Hepatitis C Virus Production by Human Hepatocytes Dependent on Assembly and Secretion of Very Low Density Lipoproteins
Huang, H., Sun, F., Owen, D. M., Li, W., Chen, Y., Gale, M., Jr. and Ye, J. Proc. Natl. Acad. Sci. USA 2007 104: 5848-5853 - Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feed back control of cholesterol synthesis and uptake.
Gong, Y., Lee, J., Lee, P., Goldstein, J.L., Brown, M.S. and Ye, J. Cell Metabolism 2006 3 15-24 - Proteasomal degradation of ubiquitinated Insig proteins is determined by serine residues flanking ubiquitinated lysines.
Lee, J., Gong, Y., Zhang, X., and Ye, J. Proc. Natl. Acad. Sci. USA. 2006 103 4958-4963 - Identification of FBL2 as a geranylgeranylated cellular protein required for Hepatitis C virus replication.
Wang C., Hua, H., Gale, M., Brown, M.S., Goldstein, J.L., and Ye, J. Mol. Cell 2005 18 425-434
Contact Us
Email
Phone: 214-648-3461
Mailing Address
Department of Molecular Genetics
UT Southwestern Medical Center
5323 Harry Hines Blvd.
Dallas, TX 75390-9046
Physical Address
L5.224