Our lab’s goals
The goal our lab’s research is to develop approaches to treat genetic neurological disorders, specifically targeting glycogen metabolism-associated diseases, such as adult polyglucosan body disease (APBD) and Lafora disease (LD), but also going beyond. Our work is also focusing on furthering the understanding of the mechanistic basis of glycogen metabolism and in particular in diseases, where insoluble glycogen is accumulating and causing neurological disease.
What’s so special about glycogen?
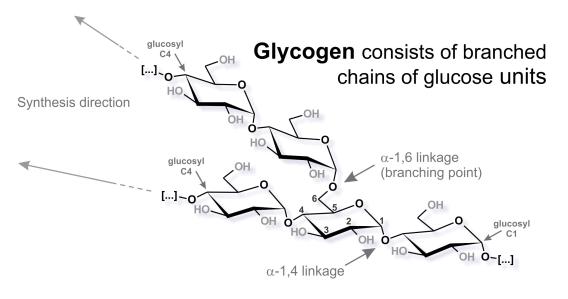
Almost all cells on this earth store energy in form of polyglucans (polymers of glucose). While animals, fungi, and bacteria store water-soluble glycogen, plants usually store water-insoluble starch. The chemical basis is the same: branched chains of glucose units. In mammalia the role of glycogen really depends on the specific organ, tissue, and even cell type. Both liver and muscle use glycogen to store excess blood-glucose after a meal. While the liver releases glycogen-derived glucose to the blood to maintain physiological blood-glucose levels, the muscle uses the glycogen for local replenishment of ATP pools after short term high-amplitude exercise. The role of glycogen in the brain is still under investigation, but it seems to be crucial during learning and in certain stress conditions.
When things go wrong: glycogen storage diseases (GSD)
Glycogen is synthesized using UDP-glucose and through the concerted action of glycogen synthase (GYS) and glycogen branching enzyme (GBE1). It is degraded by glycogen phosphorylase (GP) and the glycogen debranching enzyme (AGL) or, in the lysosome, by acid glucosidase (GAA). Many more enzymes are implicated in glycogen synthesis, and for almost all of them there is a glycogen storage disease. Some of the diseases are very severe.
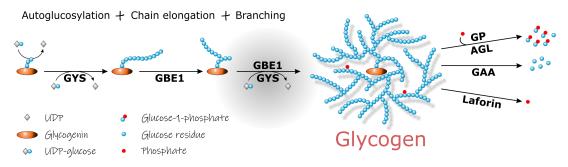
GSDs with accumulation of insoluble glycogen
In several GSDs glycogen precipitates (i.e. it becomes insoluble) and aggregates into polyglucosan bodies. These polyglucosan bodies are the hallmark of Lafora disease (LD) and adult polyglucosan body disease (APBD).
LD is a progressive, neurodegenerative myoclonic epilepsy with onset in previously healthy teenagers and death within 10 years of the first symptoms. LD Patients are lacking either of the two enzymes Laforin or Malin. Laforin has been identified as a glycogen phosphatase, removing phosphate from glycogen. Malin is an ubiquitin E3 ligase with a long list of proposed targets. One function that puts malin in the context of glycogen metabolism is the action of malin in complex with laforin to regulate the glycogen chain elongating enzyme GYS.
APBD is also a progressive neurological disorder with involvement of the central and peripheral nervous system. With onset around age 50, symptoms include neurogenic bladder, distal sensory deficits, gait difficulties, and cognitive impairment. APBD progression leads to wheelchair dependence and shortened life expectancy. Autosomal recessive mutations in the GBE1 gene are the genetic cause of APBD and the reason for accumulation of insoluble glycogen.
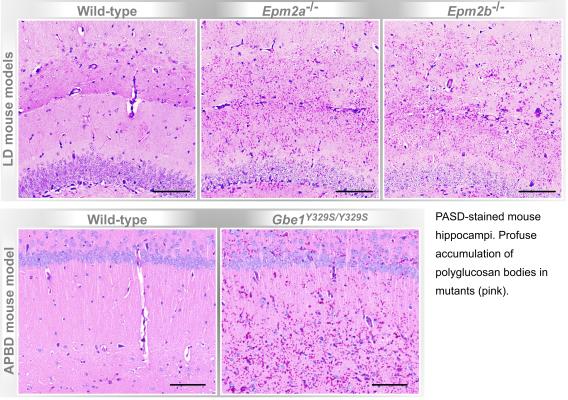 We study the mouse models of LD and APBD and of other GSDs.
We study the mouse models of LD and APBD and of other GSDs. A couple of things we found out…
The fact that laforin is a glycogen phosphatase put glycogen phosphate into the focus of LD research. We showed that glycogen is phosphorylated at the glucosyl carbons C2, C3, and C6. We found that glycogen phosphate is more abundant in the inner parts of the polyglucan molecules and in regions that contain longer glucan branches. Interestingly, we could also demonstrate that glycogen from LD mice consisted of a higher proportion of long glucosyl chains, pointing to a role of phosphate in specific phases of glycogen synthesis. We were the first to study glycogen phosphate and structure in the brain. We could show that hyperphosphorylation is a consequence of laforin deficiency but not the cause of glycogen precipitation and LD leading to a paradigm change in the field regarding LD pathogenesis. Furthermore, we demonstrated that in LD and APBD only insoluble glycogen contains abnormally long glucan chains. We could also show that normal soluble glycogen consists of molecules that differ in their propensity to precipitate as well as their content of longer glucan chains. Glycogen precipitation in LD and APBD affects only a small proportion of the soluble glycogen pool and the factors that make that glycogen precipitate are still elusive.
Going beyond…
Our research will continue to uncover the unknowns of glycogen metabolism. At the same time we are developing treatment strategies for neurological glycogen storage diseases that include gene therapy and that will be beneficial to diseases beyond glycogen and beyond the brain.