Inflammation is a rapid response of the body to microbial infections and tissue damage. Although inflammation protects against pathogenic assault and promotes healing, it can cause more damage than the inciting event if its magnitude and duration are not strictly controlled. Excessive and unrestrained inflammatory responses underlie various chronic inflammatory diseases, including autoimmunity and cancer.
Post-translational modifications mediated by ubiquitin conjugation play a critical regulatory role in immune cells. We have demonstrated earlier that the ubiquitin pathway proteins regulate T-cell tolerance and the deficiency of ubiquitin ligase Cbl-b, Itch and GRAIL cause defects in T-cell tolerance resulting in pathologic inflammation. This led to the concept that these ligases are dominant ‘toloregenic factors’ and can be targeted in inflammatory diseases and cancer immunotherapy.
The ubiquitin signal is interpreted on the basis of the number of attached ubiquitin chains and the topology of their linkage. A polyubiquitin chain is formed when one of the seven lysine residues in ubiquitin is linked to the C-terminal glycine of another ubiquitin. Although ubiquitin contains seven lysine residues, linkage generally occurs via either Lys48 (K48) or Lys63 (K63). K48-linked polyubiquitination mainly targets proteins for proteasomal degradation, whereas K63-linked polyubiquitination leads to nonproteasomal modifications, such as subcellular localization or protein-protein interactions. Similar to phosphorylation, protein ubiquitination is reversible, and removal of ubiquitin molecules is mediated by deubiquitinating enzymes such as Cyld.
We have shown that the ubiquitin ligase Itch forms a complex with Cyld that sequentially removes K63-linked ubiquitin chains and catalyzes K48-linked chains on Tak1, which is essential for attenuation of inflammatory signaling. Deficiency of Itch leads to severe lung cancer which was associated with elevated levels of tumor promoting cytokines such and IL-6 by the tumor associated macrophages. Our results further demonstrated that Itch regulates the expression of pro-inflammatory cytokines by modulating p38a phosphorylation. The pathologic consequences of losing this system in humans was highlighted by identification of a truncating mutation in Itch gene among Amish families which results in multi-organ inflammatory disorders.
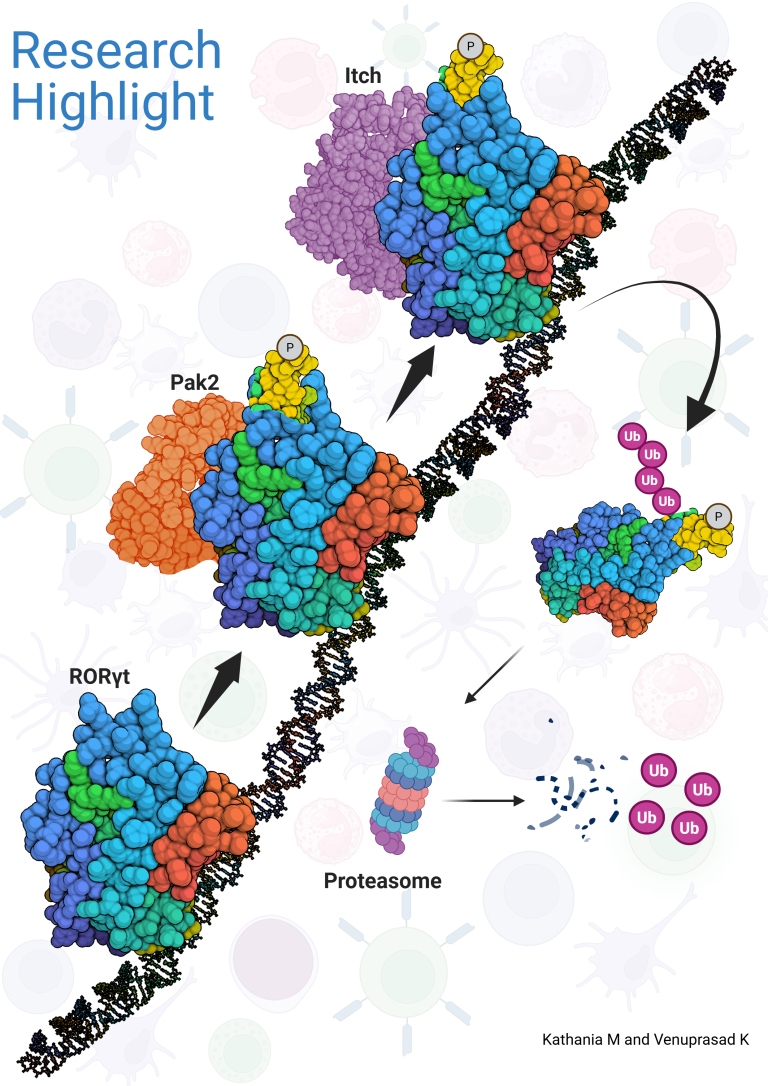
The genetic deletion of Itch in mice leads to spontaneous colitis and increased growth of colon cancer. Mechanistically, we showed that Itch recognizes a conserved ‘PPXY’ motif within the transcription factor ROR-gt and regulates IL-17 expression. We further demonstrated that defects in the enzymatic activity of Itch in ulcerative colitis patients leads to elevated IL-17-mediated colonic inflammation and fibrosis. Furthermore, we recently showed that ROR-gt function is also regulated by its SUMOylation.
We are now taking a bolder and more ambitious approach through a combination of systemic and biochemical approaches to discover new regulators of inflammation. Our research could lead to the development of novel therapeutic strategies for human inflammatory diseases and immunotherapy for cancer.