See Our Current Projects
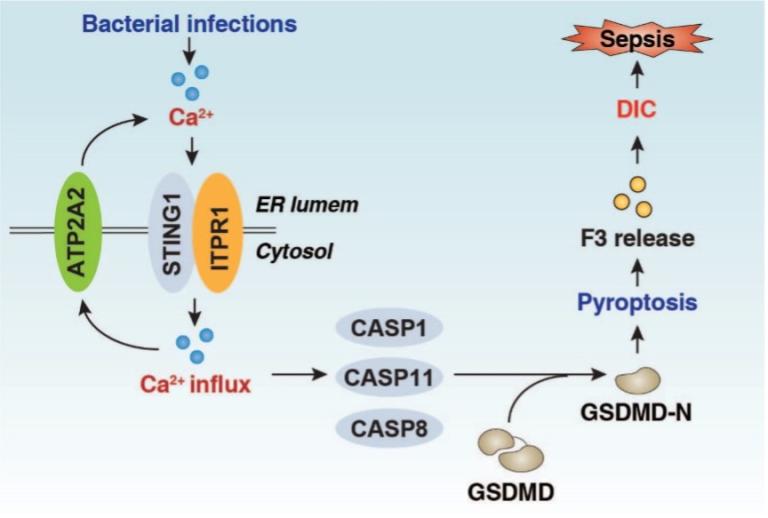
Infection-induced host immune dysfunction is associated with high mortality in the intensive care unit. Global data show that microbial infection-induced septic shock causes about 20% of deaths worldwide each year, resulting in calls for accelerated research to improve its diagnosis and treatment. The clinical course of lethal infection evolves from an early inflammation phase to a late immunosuppressive stage, in a process that is initiated by pathogen-associated molecular pattern molecules (PAMPs). A leading cause of microbial infections in hospitalized patients is Gram-negative bacteria, which release the cell wall component and PAMP lipopolysaccharide that activates immune pathways. The innate immune system is the first line of defense against invading pathogens, and excessive activation of innate immune cells can lead to cytokine storms, multiple organ dysfunctions, and even death. This process is related to the dynamic and adaptive metabolic changes of immune cells, which regulate the onset, duration, and outcomes of inflammation. As part of this project, we are investigating the complexities of immunometabolic pathways (e.g., STING1 and ACOD1) during lethal infection.
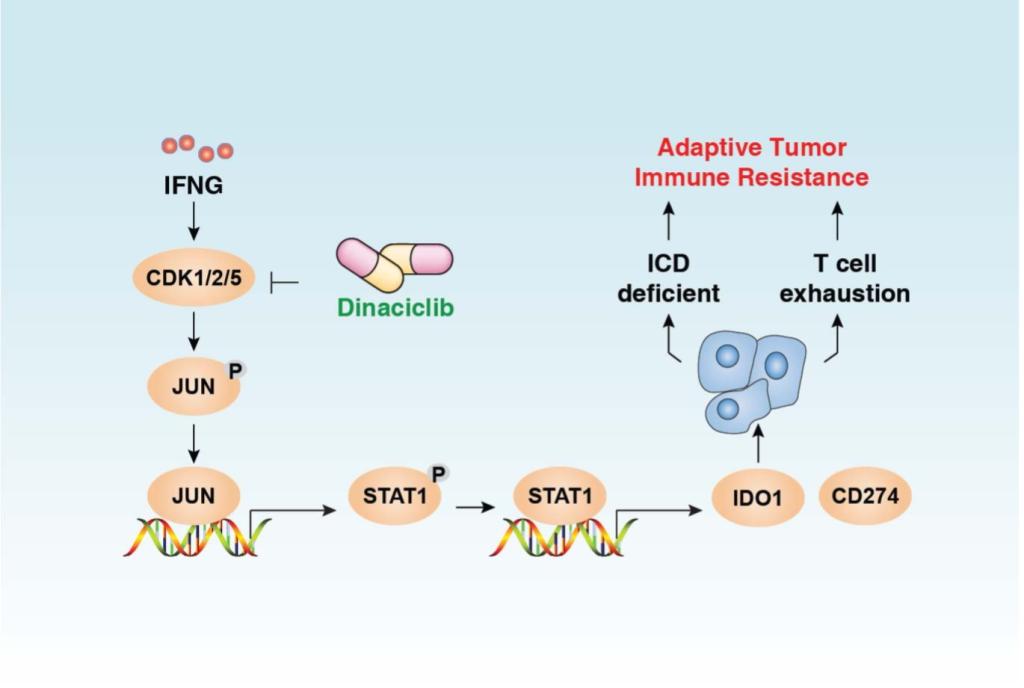
Pancreatic ductal adenocarcinoma (PDAC) is an extremely lethal cancer with limited treatment options. The application of immune checkpoint inhibitors holds great promise for improving pancreatic cancer patient outcomes, as it has already done for patients with melanoma or lung cancer. Unfortunately, to date, the use of immune checkpoint inhibitors alone or with chemotherapy in PDAC have achieved limited benefits. This is likely a result, in part, of the presence of a uniquely immunosuppressive tumor microenvironment that predominates in most human PDACs. Features of this tumor microenvironment include a highly fibrotic stroma and extensive infiltration by immunosuppressive cell populations. High stromal density can provide a barrier to the delivery of cytotoxic agents and has been postulated to limit T cell access to tumor cells and T cell function once recruited to the tumor site. Thus, agents that can overcome these stromal effects would be particularly attractive therapeutics for PDAC. As part of this project, we are investigating the mechanisms of adaptive immune resistance and the impact of iron metabolism pathways in tumor-stromal interaction.
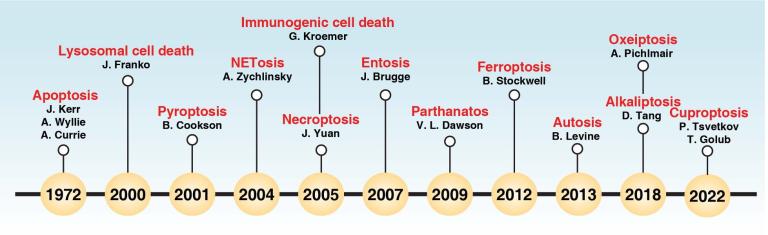
Cells may die from accidental cell death (ACD) or regulated cell death (RCD). ACD is a biologically uncontrolled process, whereas RCD involves tightly structured signaling cascades and molecularly defined effector mechanisms. A growing number of novel non-apoptotic forms of RCD have been identified and are increasingly being implicated in various human pathologies. Damage-associated molecular patterns (DAMPs) are endogenous danger molecules that are released by dead, dying or stressed cells and activate the innate immune system by interacting with pattern recognition receptors (PRRs). We are interested in defining the mechanisms of pyroptosis, alkaliptosis,and ferroptosis, as well as the pathological roles of DAMPs (HMGB1, SQSTM1, and DCN) and their receptors (AGER and TLR) in sterile inflammation and infection.
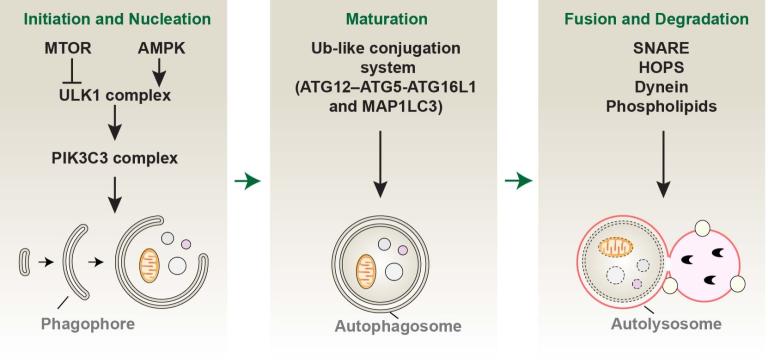
Macroautophagy (hereafter referred to as autophagy) is a conserved degradation and recycling system in organisms driven by autophagy-related (ATG) proteins and their partners. It is a dynamic process associated with the formation of membrane structures, such as phagophores, autophagosomes, and autolysosomes. Selective autophagy serves as an intracellular quality control mechanism to mediate the degradation of specific targets, such as invading pathogens, aggregated proteins, and damaged organelles. Mechanistically, specific autophagy receptors are used for cargo recognition and degradation, and thus better fulfill the catabolic necessities of the cell in response to various types of stress, including cell death stimuli. We are interested in understanding how autophagy selectivity contributes to cell survival or cell death by degrading different substrates.
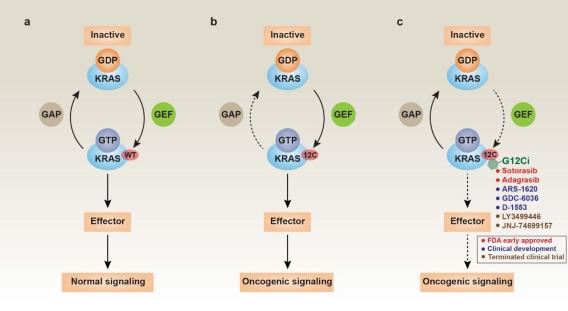
Across a broad range of human cancers, gain-of-function mutations in RAS genes (HRAS, NRAS, and KRAS) lead to constitutive activity of oncoproteins responsible for tumorigenesis and cancer progression. The targeting of RAS with drugs is challenging because RAS lacks classic and tractable drug binding sites. Over the past 30 years, this perception has led to the pursuit of indirect routes for targeting RAS expression, processing, upstream regulators, or downstream effectors. After the discovery that the KRAS-G12C variant contains a druggable pocket below the switch-II loop region, it has become possible to design irreversible covalent inhibitors for the variant with improved potency, selectivity and bioavailability. Two such inhibitors, sotorasib (AMG 510) and adagrasib (MRTX849), were recently approved for the treatment of non-small cell lung cancer with KRAS-G12C mutations, heralding a new era of precision oncology. Emerging research shows that other host point mutations in KRAS can also be directly targeted by small-molecule compounds. Recently, a novel noncovalent KRAS-G12D inhibitor, MRTX1133, showed significant preclinical antitumor activity in KRAS-G12D-bearing tumor cells, especially pancreatic ductal adenocarcinoma. We are interested in understanding how to overcome resistance to KRAS-G12C or KRAS-G12D inhibitors.