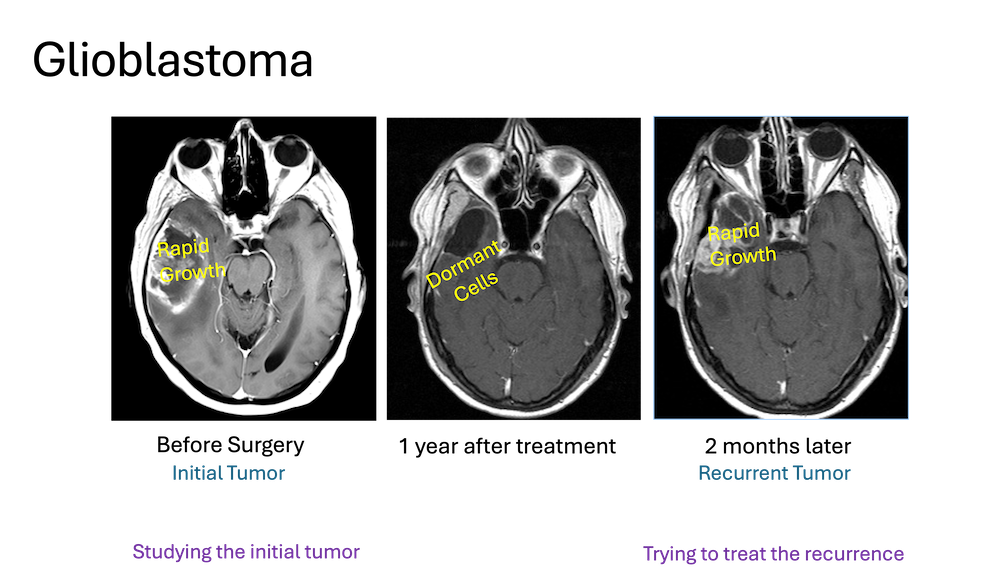
Several large-scale sequencing efforts (e.g., The Cancer Genome Atlas, NCI) and now Next Generation Sequencing (NGS), which is now consistently performed on newly diagnosed GBM tumors have the goal of identifying genetic mutations that may be matched with specific drugs at the time of recurrence after standard therapy. While ‘actionable’ mutations are often identified (e.g., EGFR, cMET, PDGFRa, CDK4, MDM2, FGFR3-TAC3, BRAF) there is little evidence that this information is of treatment benefit to individual patients (with the exception of subtype, pleomorphic xanthroastrocytoma (PXA) with a BRAF mutation). An important caveat to the disappointing results of genetic testing is the fact that NGS is performed on surgically resected tissue, demarcated by regions of MRI-T1W contrast-enhancement (CE). However, the predominant site of recurrence is in regions of non-contrast enhancement (NE) adjacent to the resection margin (80% < 3cm), which appears as a heterogenous high-intensity signal on T2W MRI. This predominant site of GBM recurrence, despite being within the radiation field, is understudied because of low abundance of the tumor cells admixed amongst normal brain stroma. Standard NGS platforms have systemic error rates are preclude them from detecting allele variants at low frequencies, which contributes to the paucity of the information on this critically important spatial region of GBM.
Image
To fill this critical knowledge gap, we established a multidisciplinary collaboration (neurosurgery, neuropathology and neuroradiology) to obtain biopsies from both contrast enhancing (CE) tumor core and non-enhancing (NE) resection margins regions using an intraoperative surgical navigation system. We sought to develop a comprehensive, multi-omic GBM Margin Atlas that could be used for understanding residual and recurrent disease as well as identify targets for treatment. We have generated a detailed multi-omic landscape of the NE region that includes bulk and single cell RNA-sequencing, FISH, metabolomics (GC-MS with C13-metabolic tracers and LC-mass spectrometry) and proteomics. We have shown that the molecular distinctions between the CE and NE regions are the result of differences in both the phenotype of GBM cells and cellular composition of the CE and NE regions. Our scRNA-Seq data suggests that GBM cells can exist along a continuum of transcriptional and phenotypic states, with non-cycling or quiescent cells representing the dominant phenotype in the NE margin. The slow cycling or quiescent tumor cell population is inherently resistant to conventional chemotherapies and radiation which may explain why the NE margin region is the site of eventual recurrence, despite receiving the highest dose of radiation and concurrent chemotherapy; these cells can be defined as being dormant (defined as a reversible phase of cell arrest). Physiological metabolic studies (13C-glucose tracer infusion) have demonstrated that despite to paucity of tumor cells, the metabolic profile is notable for a high glycolytic state, with lactate levels comparable to the tumor core regions. In collaboration with the Chung Lab (Simmons Comprehensive Cancer Center), we have adopted a Unique Dual Indexing (UDI, Illumina) strategy during the cDNA library preparation which enables an ultra-sensitive method to detect mutant alleles at low frequencies (0.1-1%) with high confidence. Current research in the lab using this technology has enabled us for the first time to generate a quantitative estimate of tumor cell frequency that is left at the tumor resection margin following a complete resection.
To date we have analyzed mutant allele frequency from the NE regions of 25 GBM have undergone resection at CUH. In current research, these patients are being prospectively followed to see if the residual tumor burden is correlated with time to local progression at surgical margin. Our proposed GBM Margin Atlas would directly inform the current debate regarding whether the extent of surgical resection should be limited to the CE region (total resection) or be extended to include the NE T2W-Flair regions.
Clinically actionable mutations are rare among IDH-wild type GBM patients (<2-5%) and there are few biomarkers (with the exception for the methylation status of MGMT) that can guide choice of chemotherapy. This grim prospect exists even though approximately 50% of GBM tumors express mutant oncogenes (including, EGFR, PDGFRa, cMET, PIK3CA, ALK-mutant fusion, CDK4, etc.) for which there are multiple small molecule inhibitors that have demonstrated remarkable efficacy for non-CNS solid tumors. At present, it is unclear if conventional or targeted chemotherapies fail in GBM because of subpopulation(s) of pre-existing drug-resistant cells (inherent resistance) and/or because GBM cells can rapidly acquire resistance (acquired resistance) following drug exposure. An additional confounding factor, based on published scRNA-Seq data, suggests that GBM cells can exist along a continuum of phenotypic states, including subpopulations that are actively proliferating, others which are slow-cycling or dormant and yet others which are non-dividing but migrating as single cells through the brain parenchyma. These phenotypic states are not immutable, but rather represent transient cellular states that GBM cells adopt in response to their local physical and/or chemical microenvironment. It is well known that slow cycling or quiescent cells are inherently resistant to conventional chemotherapies and radiation and are capable of driving tumor recurrence. We have shown that GBM cells moving through a 3D environment are highly resistant to chemotherapy and radiation. Recognition of phenotypic plasticity and intratumorally genetic heterogeneity that enables mechanisms of inherent and acquired resistance may explain, in part, why GBM has been so resistant to cancer therapies that work well in non-CNS solid tumors.
Image
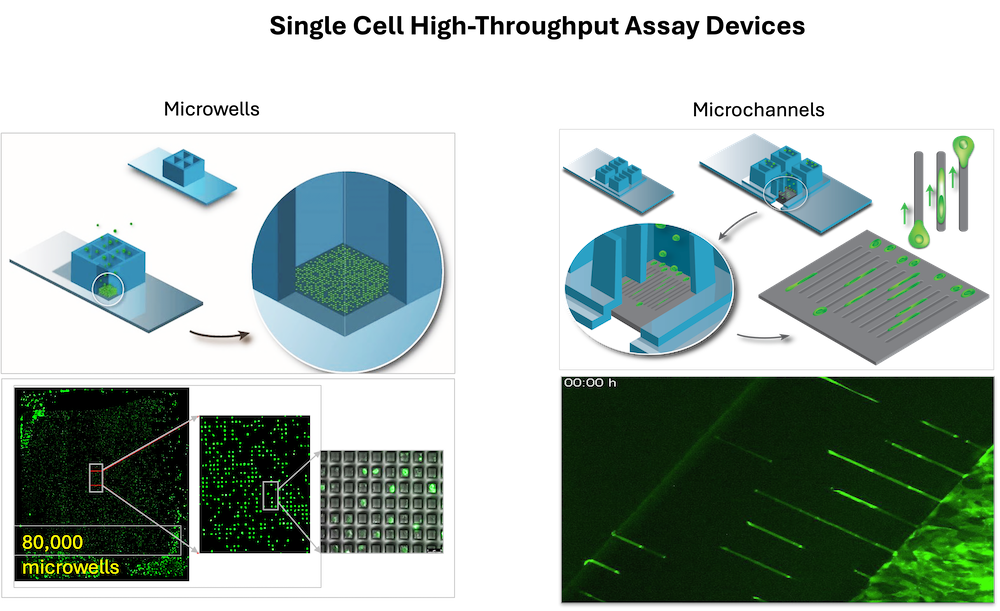
High-throughput drug screens that evaluate the ability of a compound to block or kill large populations (thousands) of rapidly dividing cells remains the cornerstone of preclinical drug development. Candidate drugs then proceed directly in vivo animal testing, toxicology screen and then early phase clinical trials, without ever assessing their impact on dormant or migrating cells. There is increasing recognition that the dormancy, migration, and CSC phenotypes represent potentially distinct but not mutually exclusive mechanisms of drug resistance that may drive recurrent disease. To address this major unmet need in preclinical drug development, we designed and fabricated the first high-throughput, large scale single-cell assay platforms for assessing drug response of tumor cells that are (i) undergoing rapid clonogenic growth, (ii) are dormant, or (iii) are migrating as single cells through a confined 3D channel. Our platform is compatible with high-content imaging and cell retrieval for downstream multi-omic profiling. The single cell and massive scale capability creates a completely new space in cancer cell drug screening. Current research is the lab uses the assay platform with a focus on direct screening of a multitude of approved drugs and chemical libraries for hits in dormant and migrating cells. The overarching goal is to find a drug or combination of drugs that can be used to kill dormant cells and stop or kill migrating cells, while using standard or newly discovered treatments to kill the proliferating cells.
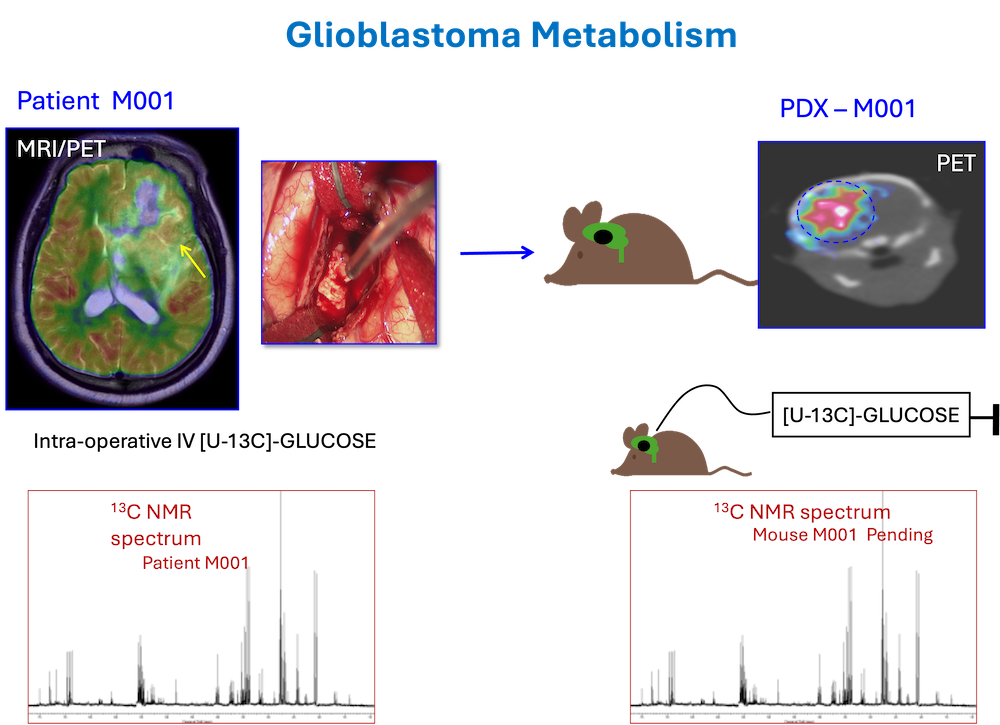
Metabolic reprogramming is a crucial adaptation of malignant transformation. Working with a multidisciplinary team (neurosurgery, anesthesiology, investigational pharmacy, and surgical nursing) our lab was the first to demonstrate feasibility and develop a protocol for intra-operative infusion of 13C-metabolic tracers in brain tumor patients undergoing surgical resection of their tumors. This protocol, which has been adapted for lung cancer, melanoma, kidney cancer, breast cancer and prostate cancer by a team of investigators at UTSW, effectively turns the operating room into an extension of the laboratory. With informed consent from patients, the tumor can be labeled with 13C-tracer (glucose, acetate, lactate, for example) and mapped ex vivo to demonstrate the critical metabolic pathways that are active in the tumor.
Our metabolic studies also extended to a repository of high-grade gliomas in Patient-Derived Xenograft (PDX) models that we developed from primary tumor samples collected at the time of the surgical procedure and injected into brains of 2-3 mice. Infusion of 13C-substrates challenged a core tenant of cancer metabolism that tumors are inflexible and ‘addicted’ to a particular substrate (glucose and/or glutamine). We have subsequently shown that GBM and brain metastases are capable of oxidizing multiple substrates, including acetate, fatty acids, and ketones.
Image
Our work raises the exciting possibility of being able to exploit a metabolic vulnerability to target cancers and/or develop more effective imaging biomarkers, both of which are urgently needed for clinical management of the patients. As an example, we hypothesize that since acetate is not an essential metabolic substrate under normal physiological conditions but is preferentially consumed relative to glucose by the citric acid cycle in cancer cells, it represents a potentially effective therapeutic target and/or imaging biomarker. Studies are ongoing in Glioblastoma, IDH-mutant lower grade gliomas, and brain metastases in the lab.
Investigate the role of acetate metabolism in gliomagenesis:
In mammalian cells, acetate can be utilized to produce acetyl-CoA in the mitochondria by ACSS1, and in the cytosol and nucleus, by ACSS2. To date, there are no genetic models to investigate the role of mitochondrial ACSS1 in cancer. Using the latest CRISPR gene-editing technology, our lab has generated novel ACSS1 and ACSS2 germline and conditional (floxed) knockout mice. These mice will be invaluable for ongoing mechanistic studies to dissect the role acetate metabolism in glioma initiation and maintenance. as well as for developing the 11C-acetate PET tracers in collaboration with Nuclear Medicine who have developed a novel clinical grade 11C-acetate PET-tracer. Glioma cells utilize metabolites, not only to support their bioenergetic and biosynthetic demands, but also for epigenetic regulation of transcriptional programs allowing tumor cells to precisely adopt to their nutrient microenvironment. An extensive panel of genetically engineered mice, (including Glut1f/f, GSf/f, HKf/f, LDHf/f, PDHf/f), when incorporated into our genetically engineered de novo mouse glioma models will allow in-depth exploration of acetate as a metabolic target in ongoing research in the lab.
Most chemotherapies and targeted therapies are developed for non-CNS tumors and are often specifically selected for poor brain penetration to limit the potential for neurotoxicity. As such, more than 95% of cancer drugs are unable to cross the blood-brain barrier (BBB) and achieve a sufficient concentration to be effective. The quest to improve BBB drug penetration has been ongoing for more than 50 years, with many strategies abandoned due to poor safety vs efficacy profiles.
Image
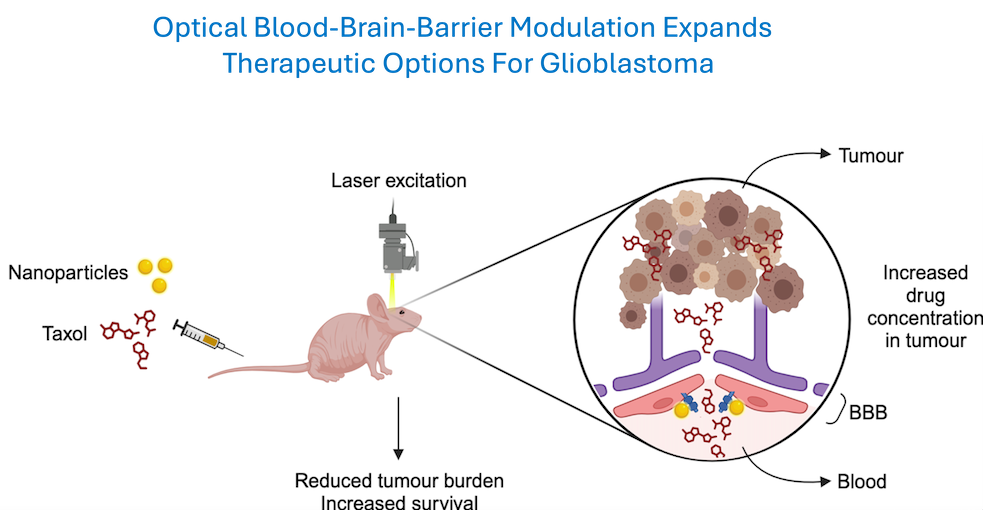
To develop a safe and effective drug delivery system across the BBB, in close collaboration with Dr. Zhenpeng Qin (UTSW/UTD School of BioMedical Engineering), an expert in optics and nanoparticle technology, we have developed a non-invasive, transcranial method of transiently disrupting the BBB. We have demonstrated that when functionalized gold nanoparticles attach to a protein forming the BBB tight-junction associated complex are activated by short-pulses of a picosecond wavelength laser light, safe and reversible opening of the BBB can be achieved with no discernable injury to the brain parenchyma. Using a novel mouse model that accurately mimics the crucial histopathological features of diffuse single cell infiltration, we recently reported that a commonly used chemotherapy (paclitaxol) that has failed clinical trials in GBM because of poor brain penetration, can achieve therapeutic levels following light activated BBB disruption and lead to a significant increase in overall survival of the mice. With funding from a recent multi-PI NIH RO1 grant, these findings are now being extended to a large animal model (pig) to evaluate feasibility for clinical translation.