CDKN2A and CDKN2B, two tumor suppressor genes within close proximity on chromosome 9p21, encode three different proteins linked to either the RB pathway (INK4A and INK4B) through CDK4/6 inhibition or the p53 pathway (ARF) by sequestering p53 antagonist, MDM2 (Fig.1). A general interest of the Skapek laboratory is to understand the transcriptional regulation of CDKN2A and B in response to different biological circumstances, mainly but not limited to eye development and cancer biology.
Arf and Eye Development
A novel role of ARF in development was first identified by the Skapek laboratory, from discovering a blind eye phenotype from Arf-/- mice but not in p53-/- mice. Further investigation revealed the function of Arf in vascular regression at later states of ocular development (Fig.2). The Arf dysfunction results in catastrophic damage to the retina and the lens and mimics a congenital human eye disease known as Persistent Hyperplastic Primary Vitreous (PHPV), which was described close to 100 years ago but still lacks a clear genetic cause. Over the past years, by making and characterizing genetically engineered mouse models, our team has identified a new Tgfß2 (Transforming growth factor beta 2)-Arf-Pdgfrß(Platelet-derived growth factor receptor beta) pathway governing the action of Arf in the developing eye . In pericytes flanking certain vessels in the eye, Tgfß2- dependent Arf expression limits the expression of Pdgfrß to control the number of these pericytes. Arf appears to do so in a manner that does not depend on p53.

Without Tgfß2 or Arf, deregulated Pdgfrß drives excess proliferation of pericytes to support the underlying vasculature, leading to its persistence and the consequent ocular damage. Our team is focused on further understanding how Tgfß2 influences Arf expression and how p19ARF controls Pdgfrß.
Remote control of CDKN2A/B at upstream non-coding region
Accumulating evidence shows the existence of cis regulatory enhancers, located in a “gene desert,” that are essential for context-specific regulation of CDKN2A and B. Genome-wide association studies (GWASs) have linked the SNP clusters on chromosome 9p21.3 situated near the CDKN2A/B locus with coronary artery disease (CAD) and type 2 diabetes (T2D) (Fig.1), suggesting direct or in-direct functions of CDKN2A/B in these diseases. (Note that SNPs associated with various types of cancer are also scattered along this non-coding region.) With mice lacking the orthologous region of the so-called CAD risk interval (chr4Δ70kb/Δ70kb), members of the Skapek laboratory showed that these animals also display primary vitreous hyperplasia, and that Arf expression is low in the primary vitreous and Tgfβ fails to induce Arf in MEFs from the chr4Δ70kb/Δ70kb mice.
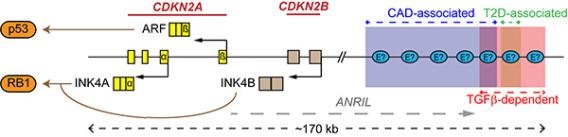
Schematic diagram showing the CDKN2A/B locus, which encodes three tumor suppressor proteins p14ARF, p16INK4A, and p15INK4B, and its associated cis enhancers.
In the setting of cancer biology, deregulation of CDKN2A/B, either by copy number loss, promoter methylation, or by disrupting normal regulatory circuits, plays an important role in cancer development. Studying the mechanisms underlying this deregulation presents entry points to further explore pharmacological strategies to restore CDKN2A/B expression in cancer. Our team has shown ARF to be the effector of TGFβ-driven cancer suppression in certain human cancer cell lines. Besides, this TGFβ-ARF/INK4B pathway depends on a de novo 20kb enhancer region engaged by TGFβ, which is absent in those TGFβ “non-responsive” human cancer lines (Fig.1). To further study the enhancer activities on CDKN2A/B regulation we have coupled novel computational methods and CRISPR technologies to focus on: 1) identifying enhancers involved in CDKN2A/B regulation in both human and mouse models, 2) understanding the molecular details regarding how remote enhancers affect CDKN2A/B regulation under different contexts, and 3) developing clinically actionable ways to restore CDKN2A/B activities.