The role of PLAG1 in muscular dystrophy
We recently discovered a new mouse model for muscular dystrophy. By crossing LSL-PLAG1 mice with MCK-Cre mice, we achieved tissue-specific expression of PLAG1 in heart and skeletal muscle. The Cre/PLAG1 mice died prematurely with evidence of cardiomyopathy and skeletal muscular dystrophy. Histology of cardiac and skeletal muscle revealed dystrophic features, including loss of muscle fiber and variation in diameter of muscle fiber, centralized nuclei, and fibrosis, which mimics human MD histology. Immunoblot revealed evidence for PI3K/AKT activation with increased expression of phospho-AKT in dystrophic muscle. RNA-Seq revealed down-regulation of DMD and other genes, abnormalities of which are implicated in forms of MD. Repression of DMD by PLAG1 was confirmed in dystrophic skeletal muscle and various cell culture models. We are currently investigating the mechanisms by which PLAG1 represses DMD and other muscular dystrophy genes.
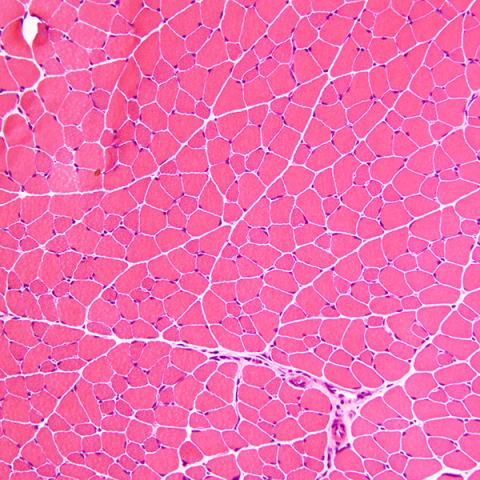 | 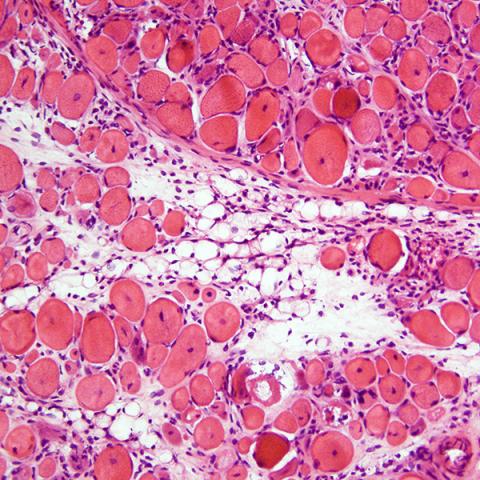 |
| H&E staining of cryo-sectioned WT | LSL-PLAG1;MCK-Cre mouse quadriceps muscle. 200x. |
Human iPSCs as a novel model to study skeletal muscle biology and RMS
The discovery of induced pluripotent stem cells (iPSCs), through reprogramming of human adult fibroblasts using the factors Oct4, Sox2, c-Myc, and Klf4, has helped propel differentiation studies and created significant advantages for the study of various developmental processes. In our lab, we derived skeletal myogenic progenitors from hiPSCs (hiPSC-MPCs) to study embryonic development of skeletal muscle, specifically the transition between a proliferating myoblast and a differentiating myocyte. We are also using this system to understand how perturbations in the normal skeletal muscle development process (ie. Copy number gains or dysregulated expression of oncogenic drivers) contributes to rhabdomyosarcoma (RMS) formation. RMS is composed of skeletal myoblast-like cells, that express early myogenic transcription factors but fail to robustly induce the full complement of muscle differentiation genes and they cannot foster cell cycle arrest. Because normal muscle differentiation is coupled to irreversible cell cycle arrest, we feel that understanding why these processes fail may eventually point toward new treatment strategies for RMS and other muscle related diseases.
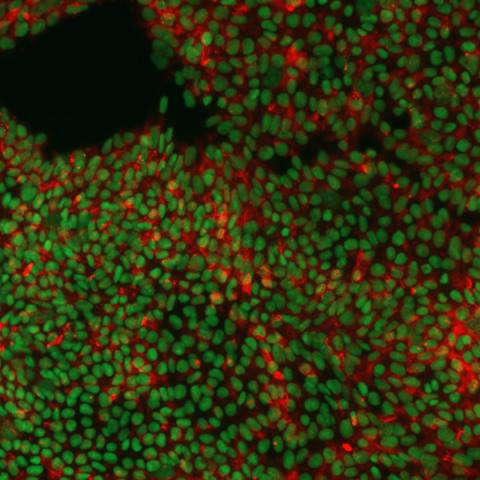 | 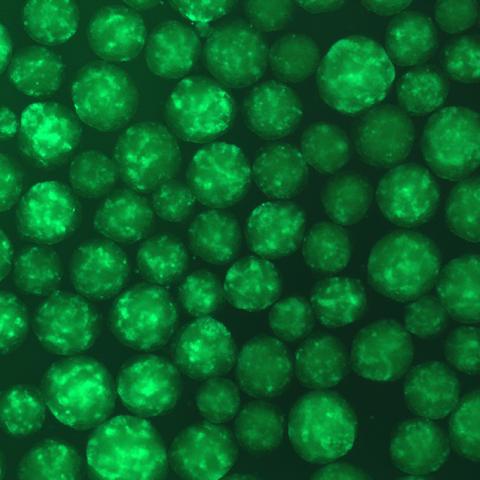 |
| Immunofluorescence image of hiPSCs stained for pluripotency markers SOX2 (green) and TRA-1-60 (red). | GFP expression in embryoid bodies (EBs) during differentiation of hiPSCs into myogenic progenitors. |
Metabolism of Translocation-driven Tumors
Another project within our lab is to understand how certain transcription factors drive changes in the metabolomics profiles of primary cell lines and patient-derived xenografts (PDXs). The PAX3-FOXO1 (P3F) fusion oncogene is sufficient to drive rhabdomyosarcoma formation and reprograms metabolism in mammalian cells. We previously utilized an estrogen receptor-based system of P3F expression in a fusion-negative rhabdomyosarcoma cell line (RD), allowing us to study how gain of P3F alters cell metabolism. In parallel, we are applying the novel auxin-inducible degron system for rapid and precise proteolytic knockdown of P3F. Both systems will allow us to obtain a detailed metabolomics profile of P3F due to their rapid timescale. Identifying unique signatures will allow us to identify key enzymes that interplay with important metabolites and candidate genes that may contribute to the metabolic reprogramming. We are also seeking to determine whether we can identify the metabolic profiles from RD and RH30 cell lines to determine if they have similar prognostic values in fusion-positive and –negative tumors.