One major focus of our research is the catalytic asymmetric [2,3]-rearrangements of reactive zwitterions, which are synthetically versatile molecules implicated in a diverse range of chemical transformations. These reactive substrates were traditionally thought to be incompatible with enantioselective catalysis because of their propensity to undergo facile thermal [2,3]-rearrangements in the absence of a catalyst. Our group has challenged this assumption held widely in our field for decades.
![Metal-Catalyzed [2,3]-Rearrangements](/sites/default/files/styles/coh_small/public/2022-05/Asymmetric-rearrangements-RS.jpeg?itok=oEuaJ6A3) Metal-Catalyzed [2,3]-Rearrangements
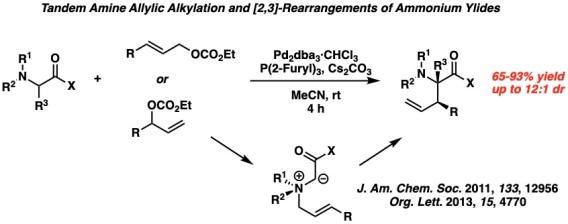
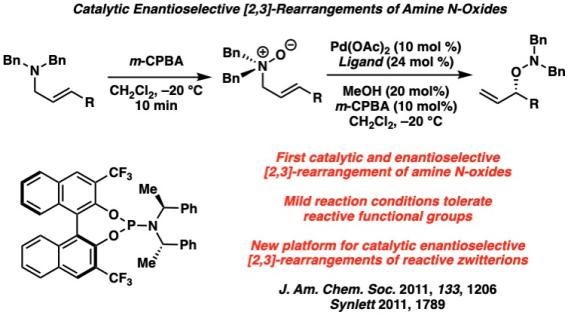
Metal-Catalyzed [2,3]-Rearrangements We have developed a series of first-in-class metal-catalyzed stereoselective [2,3]-rearrangements of zwitterionic reactive intermediates, including ammonium ylides formed through a palladium-catalyzed amine allylic alkylation, allylic amine N-oxides with palladium, and a full program of rearrangements of allylic iodonium ylides.
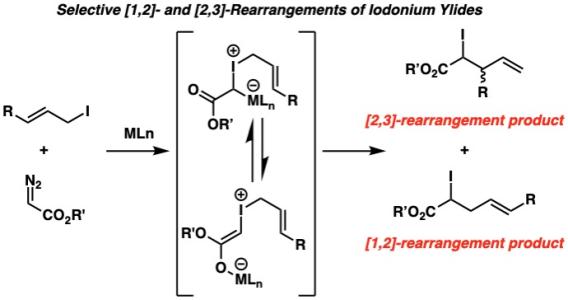
Similar to allylic ammonium ylides and allylic amine N-oxides, allylic iodonium ylides can undergo thermal [2,3]-rearrangements with the added possibility of a [1,2]-rearrangement, giving rise to sixteen possible products. Utilizing copper catalysis, we can transform iodonium ylides formed from their corresponding diazoesters and allylic iodides with copper into the [2,3]- and [1,2]-rearrangement products with high regioselectivity and diastereoselectivity. Our first investigation found that sterically hindered, electron withdrawing ligands on copper favored the [2,3]-rearrangement product, and electron donating phosphines furnished the [1,2]-rearrangement product.
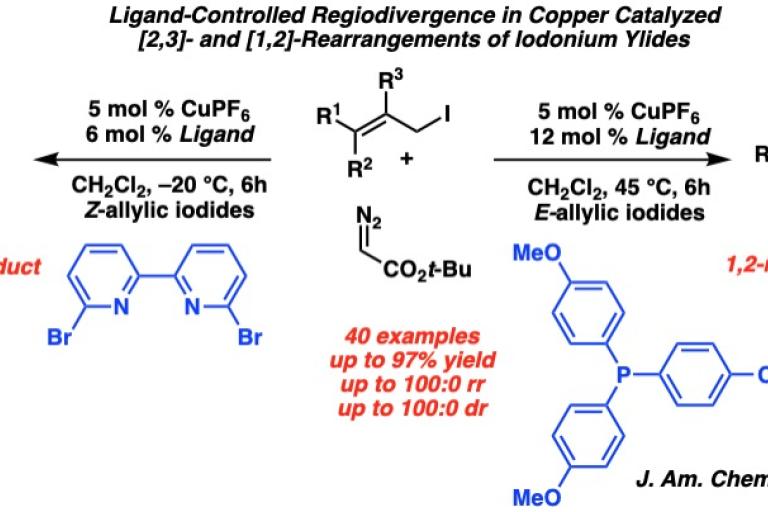
After this initial discovery, our group then sought to answer two questions in iodonium ylide rearrangements: could we alter the properties of a bipyridyl ligand to yield the [1,2] product rather than the [2,3], and could we access these rearrangement products in an enantioselective fashion?
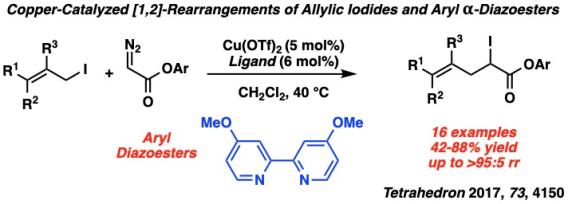
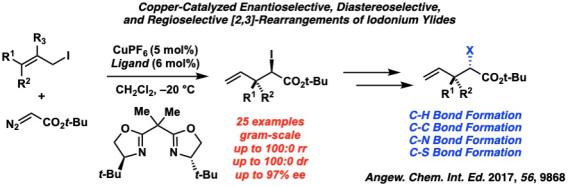
To answer these questions, we first developed the rearrangement of allylic iodonium ylides from aryl diazoesters utilizing an electron donating, non-hindering bipyridyl ligand that gave the [1,2]-rearrangement product in high regioselectivity. Furthermore, in order to access enantioenriched iodoesters, we established an enantioselective variant of the [2,3]-rearrangement with a chiral bisoxazole ligand. The carbon-iodide bond in the α-iodoester products can be functionalized with stereospecificity into C-H, C-C, C-N, and C-S bonds to give highly substituted chiral products.
Phosphoric Acid Catalyzed Rearrangements
The aromatic aza-[3,3]-Claisen rearrangement has great potential for generating medicinally valuable aromatic amines. Despite the development of several catalytic enantioselective aliphatic aza-Claisen rearrangements in recent years, the asymmetric catalysis of aromatic aza-Claisen rearrangements remains an underdeveloped area. The paucity of enantioselective aromatic aza-Claisen rearrangements may be partially attributed to the absence of a secondary interaction for two-point binding in an aromatic amine that only contains one basic functional group. Given the prevalence of indoles in biologically active molecules, we have developed an enantioselective Brønsted acid catalyzed [3,3]-rearrangement around the indole scaffold.
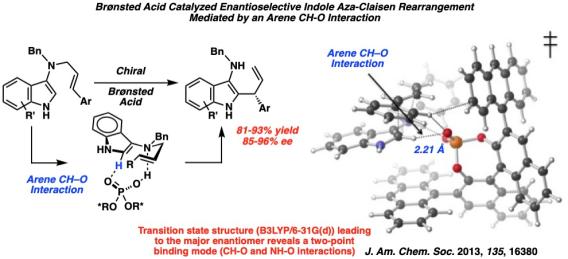
In collaboration with Professor Dean Tantillo, a computational chemist at University of California, Davis, we uncovered evidence for an arene CH-O interaction as a source of activation and stereoinduction in the reaction. Our discovery establishes a new paradigm for two-point interactions in catalytic enantioselective reactions of substrates with only one basic functional group. We anticipate that this weak interaction will be useful in the design of other Brønsted acid catalyzed enantioselective reactions that were previously thought to lack a mode for high stereoinduction through one-point binding.
Stereoselective Functionalization of Hydrocarbons
Our group has explored enantioselective rearrangements for the stereoselective functionalization of hydrocarbons found in inexpensive and abundant components of petrochemical feedstock. Terminal alkenes react with sulfur-based oxidants developed in our laboratory to generate a zwitterionic species, which we have then engaged in variety of processes. This zwitterionic species can undergo a [2,3]-rearrangement to access the allylic amination product or nucleophilic displacement for formal alkylation of an allylic C-H bond with Grignard reagents.
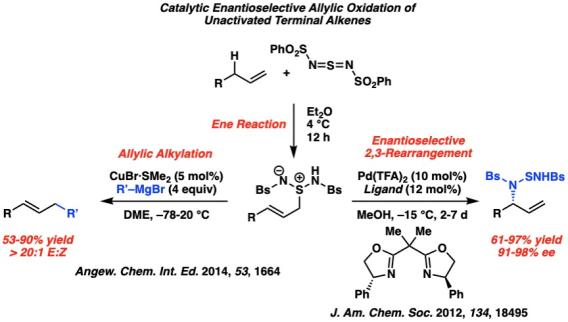
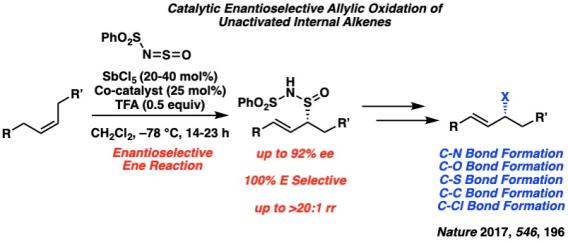
As an extension of our approach to the catalytic allylic functionalization of unactivated terminal olefins, we have recently solved the more challenging problem of catalytic asymmetric allylic functionalization of internal olefins. While most strategies for allylic oxidation of olefins are based on metal-catalyzed C–H activations and C–H insertions, these processes are inherently non-regioselective for the oxidation of unactivated internal olefins in the absence of directing groups.
We hypothesized that if the hetero-ene reaction could be rendered stereoselective by a chiral catalyst, this reaction manifold may provide the ideal platform to develop a highly enantioselective, E/Z selective, and regioselective allylic oxidation of unactivated internal olefins. This approach was successfully realized with a SbCl5-BINOL catalyst system. This method represents the first example of selectively converting unsymmetrical internal olefins into allylic functionalized products with high stereoselectivity and regioselectivity. Stereospecific transformations of the multifunctional allylic oxidation products highlight the potential for rapidly converting internal olefins into a broad range of enantioenriched structures that can be utilized in the synthesis of complex target molecules.
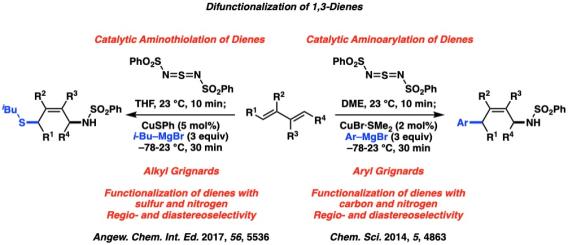
We have also utilized sulfur oxidants to selectively functionalize 1,3-dienes, which react via a [4+2] cycloaddition to generate a cyclic sulfonamide. This reactive intermediate can be treated with a copper catalyst and either aryl Grignard reagents to afford the 1,4-aminoarylation products, or alkyl Grignard reagents to afford the 1-4 aminothiolation products. Despite the importance of sulfur containing chemical structures in medicinal chemistry and material science, the synthesis of multi-functional sulfur-containing molecules still remains a significant challenge.
Current efforts in catalysis in our laboratory include the development of rearrangements of a variety of in-situ generated ylides, selective allylic functionalizations, and further uses of sulfur-based oxidants.