Our lab focuses on preclinical in vitro and in vivo studies to show how targeted radiation therapy (RT) alone or in combination with various therapeutic agents triggers an immune response. The information from these studies will pave the way for more testing of this therapeutic combination in clinical trials and translational studies to benefit patients with cancer. The studies completed thus far have offered valuable insights into the role of the innate immune system in mediating tumor cell killing as a result of targeted radiation leading to sterile inflammation. Our studies have also prompted us to exploit the potential of a novel drug delivery platform to release anti-cancer agents upon activation by RT at the tumor target and its prospect to generate anti-tumor immune responses. Emerging topics such as the effect of heavy particle radiation on the immune system as a mode of cancer therapy are also a major focus of studies. Future studies on these topics will help us to understand the mechanisms related to immune-mediated tumor inhibition by combined RT and immunotherapy.
The successful combination of immunotherapy and radiation therapy (RT) depends on a better understanding of how RT modulates the tumor microenvironment, especially tumor-infiltrating leukocytes (TILs). Using multiple syngeneic mouse tumor models, we recently reported (Takeshima et al, PNAS, 2016) that RT induces a rapid infiltration of CD11b+Gr-1high neutrophils into tumors within 24 to 48 hours (Fig 1). These RT-recruited tumor-associated neutrophils (RT-Ns) exhibit an increased production of reactive oxygen species and induce apoptosis of tumor cells. Notably, RT-Ns do not express F4/80 and are not part of myeloid-derived suppressor cells (MDSCs). Importantly, RT-Ns are at least in part responsible for the tumor inhibition effects of RT (Fig 2). Furthermore, concurrent administration of granulocyte colony-stimulating factor (G-CSF), a well-known stimulator of neutrophils, enhances the anti-tumor effects of RT (Fig 3). In addition, G-CSF enhances RT-mediated production of tumor-specific cytotoxic T lymphocytes (CTLs) in the tumor, and this effect depends on RT-Ns (Fig 4).
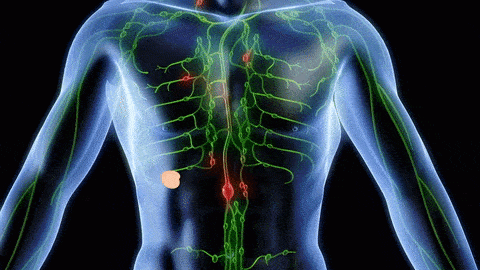
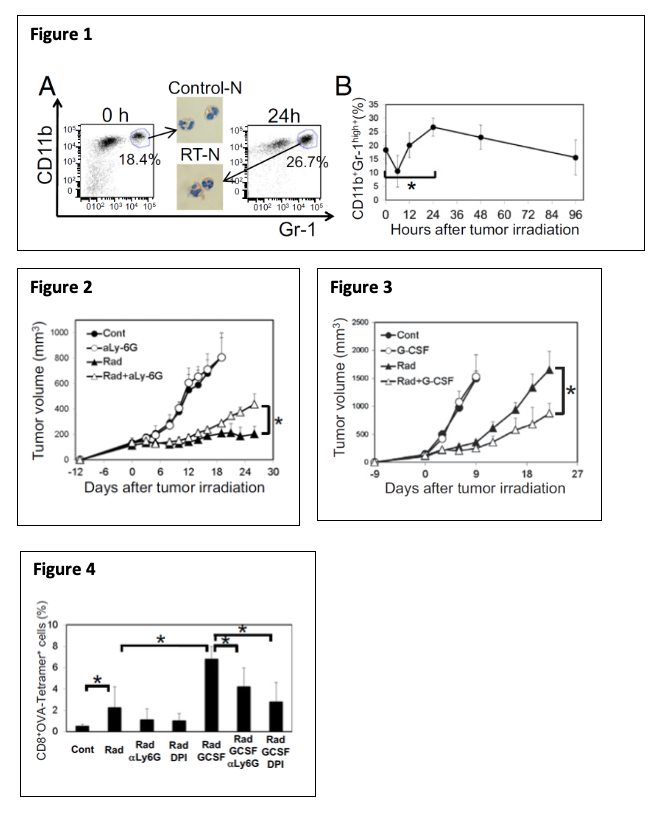
All these findings suggest that RT-Ns play an important role in RT-induced anti-tumor immunity. These newly discovered early events of RT-Ns indicate a tumor-specific response that reveals anti-tumor properties. Such innate and inflammatory responses often pave the way for more specific adaptive immune responses although their underlying mechanisms are largely unknown. Therefore, neutrophils may be the missing link to how DNA-sensing pathway activation leads to anti-tumor immunity. Exploiting this mechanism can optimize RT-induced DNA-sensing activation, leading to a more robust anti-tumor immune response and eventually overcoming tumors’ resistance to immunotherapy. Currently, we are investigating the mechanisms underlying the accumulation of RT-Ns in the tumor after RT. Specifically, we are identifying which DNA-sensing pathways and which adhesion molecules, cytokines, and chemokines as a downstream effect of DNA-sensing are responsible for recruiting RT-Ns. In addition, we hypothesize that RT-Ns is the key to enhancing DNA sensing by RT. Therefore, we are also identifying the mechanism of enhanced DNA sensing by RT-Ns. Furthermore, we are evaluating the anti-tumor effects of potentiating DNA sensing effect of RT-Ns with other drugs (e.g. G-CSF + beta-lapachone).
Radiation therapy (RT) is one of three core modalities used to treat cancer, although tumor recurrence is observed even with high doses of fractionated stereotactic ablative radiotherapy (SAbR). Recent studies have revealed that RT-induced tumor cell death is “immunogenic” because it acts like an “in situ” vaccination and triggers an antitumor immune response, including cytotoxic T lymphocytes (CTLs), the main effectors against tumor cells. The potential combination of immunotherapy and RT is based on its well-known immunogenic properties. Because RT is targeted directly to the tumor, it does not inherently have an immune-compromising effect on the host.
Sterile inflammation occurs after tissue damage in the absence of pathogens. The hallmark of sterile inflammation is the rapid infiltration of neutrophils, as observed in the spleen, thymus, and gut after whole-body ionizing radiation in mice. Inflammatory responses are triggered by damage-associated molecular patterns (DAMPs), which are released from cells dying from exposure to RT. A previous study from our lab revealed that sterile inflammation caused by RT involves tumor-associated neutrophils (TAN) that induce tumor cell killing mediated by reactive oxygen species and, eventually, by the involvement of tumor-infiltrating CTLs.
DNA sensing as a part of DAMPs is an important aspect of RT-mediated immune response. One of our ongoing projects aims at [1] investigating the involvement of RT-induced DNA sensing in RT-induced tumor neutrophilic infiltration (RT-N); [2] gaining insights into the mechanisms of enhanced DNA sensing by RT-Ns and [3] modulating RT-N to enhance DNA-sensing mediated tumor suppressive effect.
Our findings from ongoing and future experiments will provide valuable insights into the DNA-sensing pathways activated by RT, the tumor microenvironment, and radiation therapy’s interaction with the immune system for better therapeutic intervention in patients with cancer.
Natural killer (NK) cells are components of innate immunity and key effectors against tumor cells. The natural-killer group 2, member D (NKG2D) is a stimulatory receptor expressed on NK cells, NKT cells, and subsets of γδ+T cells. The ligands of NKG2D are expressed on the surface of transformed cancer cells (lung, pancreas, kidney). Radiation therapy (RT) may upregulate the expression of NKG2D ligands. In humans, these ligands were reported as major histocompatibility (MIC) A, MIC B, and UL-16 binding protein (ULBP) 1-6. The NKG2D ligands expressed on tumor cell surface upon binding to NKG2D activate NK cells. The NKG2D ligands expressed on the tumor cell surface are shed by metalloproteases (abundantly present in cancer) and released as soluble forms. ULBP2, an NKG2D ligand, is expressed in various tumor tissues and released into a soluble form by sheddases, ADAM 10 and 17 (a-disintegrin and metalloproteinase 10 and 17), which are also upregulated on the tumor cell surface.
Our preliminary data show that gamma RT leads to increased expression and release of soluble sULBP2 from multiple human types of cancer cell lines into the supernatant (lung, pancreas, kidney), which may interfere with NK-mediated killing of tumor cells. Our research will determine the underlying mechanisms and avenues to manipulate them with the final goal of increasing RT’s efficacy in activating NK cells against tumor cells.
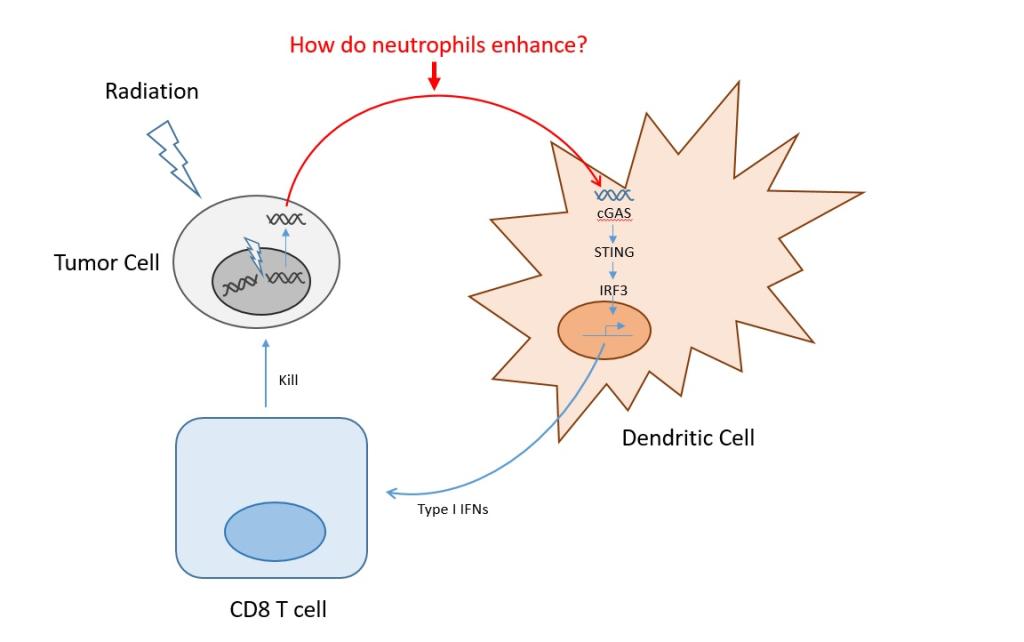
Ionizing radiation has unlimited tissue penetration and has already become a part of routine cancer therapy. Recent advances in the delivery of γ-radiation therapy (RT) such as 3-dimensional conformal (3-DCRT), intensity-modulated (IMRT), image-guided (IGRT), and stereotactic (i.e. SAbR, CyberKnife, Gamma Knife, etc.) radiosurgery allow for highly localized dosing within tumor tissue anywhere in the body, while sparing the surrounding healthy tissue. When combined, these features make targeted drug release and/or activation using ionizing radiation highly tractable and desirable. Targeted activation of therapeutics by RT is still a relatively nascent field. In addition, the avenue of a synergistic combination of these activated therapeutics with the body’s immune response to radiotherapy is still unknown. To that end, we have been investigating drug and RNA release from a non-infectious virus-like particle (VLP) called Qβ and found that RT can trigger the release of the clinically relevant drug doxorubicin, as well as shed fragments of RNA, which we believe will synergistically generate a local immune response in the tumor environment.
Our central hypothesis is that optimized macromolecular single-stranded RNA can be degraded under very low doses of RT to release small immunogenic strands of RNA as well as small molecule drugs from the inside of a porous VLP capsid that is linked to the capsid via tRNA.
This ongoing project is essential because it exploits advancing technologies on focused RT to create nanoscopic drug carriers that can be activated at any tissue depth. Further, ionizing radiation is being applied for the first time to activate RNA degradation for drug release from a novel drug delivery tool and has been used for a potential role in immunomodulation.
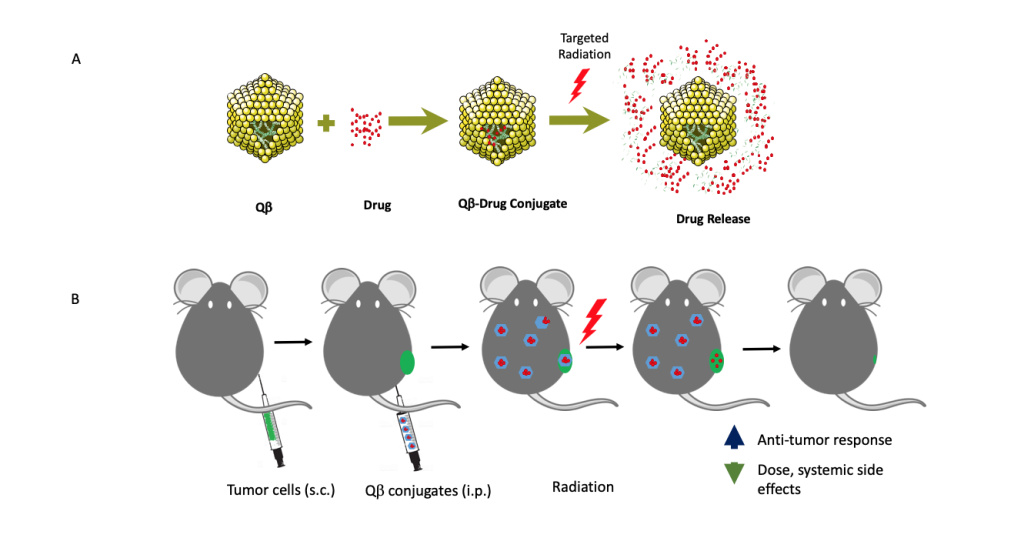 A proposed model of targeted radiation-induced therapy release from virus-like-drug delivery platform Qb. (A) 180 Qb protein subunits self-assemble around random E. Coli RNA that conjugate with therapeutic agents. Radiation releases these agents from Qb. (B) In vivo release of therapeutic agents from Qb upon targeted radiation.
A proposed model of targeted radiation-induced therapy release from virus-like-drug delivery platform Qb. (A) 180 Qb protein subunits self-assemble around random E. Coli RNA that conjugate with therapeutic agents. Radiation releases these agents from Qb. (B) In vivo release of therapeutic agents from Qb upon targeted radiation. In vivo studies have shown that radiation therapy (RT) induces the release of damage-associated molecular patterns (e.g. HMGB1, calreticulin) into the extracellular matrix, spurring the recruitment and activation of antigen-presenting cells, which then prime cytotoxic T cells. Thus, RT works synergistically with immune activation mediating tumor death. Recent studies have confirmed that more HMGB1 is released with carbon ion RT than with gamma RT. In addition, evidence suggests increased clustered DNA damage producing higher levels of irreparable DNA fragments that elicit a higher innate immune response, upon reaching the cytosol, after carbon ion RT.
RT can potentially affect pancreatic cancer (PaCa), one of the deadliest human cancer types, as an adjuvant treatment for chemo- and immunotherapy. However, PaCa cells use complex immune escape strategies to circumvent T-cell recognition, including activation/upregulation of immune checkpoint molecules, downregulation of classical MHC I expression, and upregulation of HLA-E and HLA-G molecules, or intratumoral recruitment of T regulatory and myeloid-derived suppressor cells. Our ongoing research aims to determine whether carbon ion RT can initiate immune/inflammatory changes in human and murine PaCa cell lines that can specifically promote tumor sensitization.
We are also interested in studying the changes in tumor mutation burden (a measurement of induced neoantigens) in human and mouse PaCa cell lines mediated by carbon ion RT. Evaluating neoantigens can define potential new targets for immunotherapy (targeted antibody therapy, checkpoint inhibitors, vaccines) and identify tumor predictor markers for PaCa therapies. Our strategy to quantify the changes in RT-mediated neoantigens involves analyzing the whole exome sequence data and whole transcriptome (RNA-seq) data of each cell line before and after RT. Then, multiple in silico bioinformatics prediction algorithms are used to identify the kinetics/de novo generation of potential MHC-I restricted neoepitopes.