The Orth Lab is interested in elucidating the activity of virulence factors from pathogenic bacteria. We aim to gain novel molecular insight into the virulence mechanisms caused by effectors as well as the eukaryotic signaling systems they emulate or target.
Many virulence factors are secreted by bacteria using a type III secretion system (T3SS). The T3SS resembles a needle-like structure that efficiently ejects effector proteins from bacteria into the cytosol of a host cell. Effectors have evolved in a manner similar to many viral oncogenes; a eukaryotic activity is usurped and modified by the pathogen for its own advantage.
We are working on bacterial T3SS systems, their regulators and their secreted effectors to understand how signaling systems in the eukaryotic host can be manipulated by bacterial pathogens. These studies provide novel insight into the molecular workings of eukaryotic signal transduction. Our work at UT Southwestern is accomplished using a broad range of tools, including biochemistry, molecular microbiology, protein chemistry, structural biology, yeast genetics, cell biology, and more.
Orth Lab Research Highlights in Vibrio
We study the virulence mechanisms of Vibrio parahaemolyticus (Vpara, pictured above using confocal microscopy, infecting HeLa cells). Vpara is a Gram-negative bacterium commonly found in marine and estuarine environments. Infection typically results from consumption of contaminated shellfish and leads to acute gastroenteritis. Individuals who are immunocompromised or burdened with pre-existing health conditions are at high risk for severe, potentially lethal complications. The Vpara genome contains two pathogenicity island - encoded type III secretion systems (T3SS1 and T3SS2), with corresponding effectors (Vops) that contribute to its pathogenicity. T3SS2 transcription is regulated by a co-component signal transduction system VtrA/VtrC that senses bile acids in the host gut. VtrA/VtrC is differentially activated by microbiome modified bile acids TDC (activator) and CDC (antagonist), opening the possibility of Vpara pathogenicity being influenced by an individual's microbiome.
Vibrio T3SS1
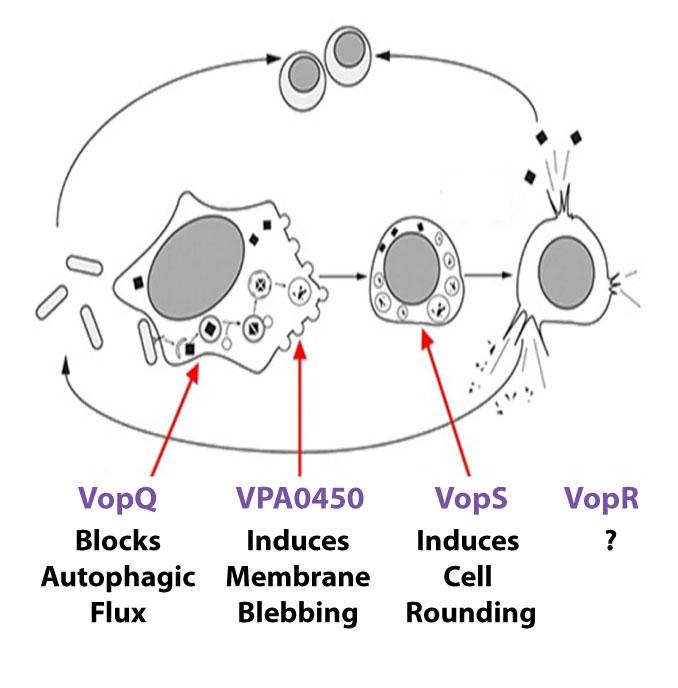
Our studies demonstrate Vpara uses T3SS1 to orchestrate host cell death by autophagy induction, cell blebbing, cell rounding, and then cell lysis. We found deleting one effector, VopQ, completely abolished autophagy induction in infected cells. The next step entails collapse of the actin cytoskeleton with synchronized rounding of infected cells. We attributed the cell rounding to VopS, an effector that AMPylates the Rho family of GTPases, thereby preventing them from interacting with downstream signaling molecules. Another T3SS1 effector VPA0450 induces membrane blebbing and facilitate host cell lysis via its inositol polyphosphate 5-phosphatase activity that disrupts the dynamic interaction of the cell membrane with the actin cytoskeleton. Many other effectors are thought to contribute to Vpara - mediated cell death. Future investigations will be directed at understanding the molecular mechanisms they employ to disrupt host cell signaling and promote infection.
Vibrio T3SS1 orchestrates host cell death
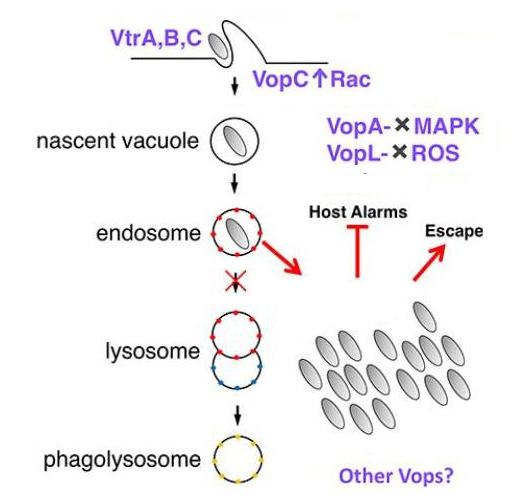
Vibrio T3SS2 establishes a protective intracellular niche
Vibrio T3SS2
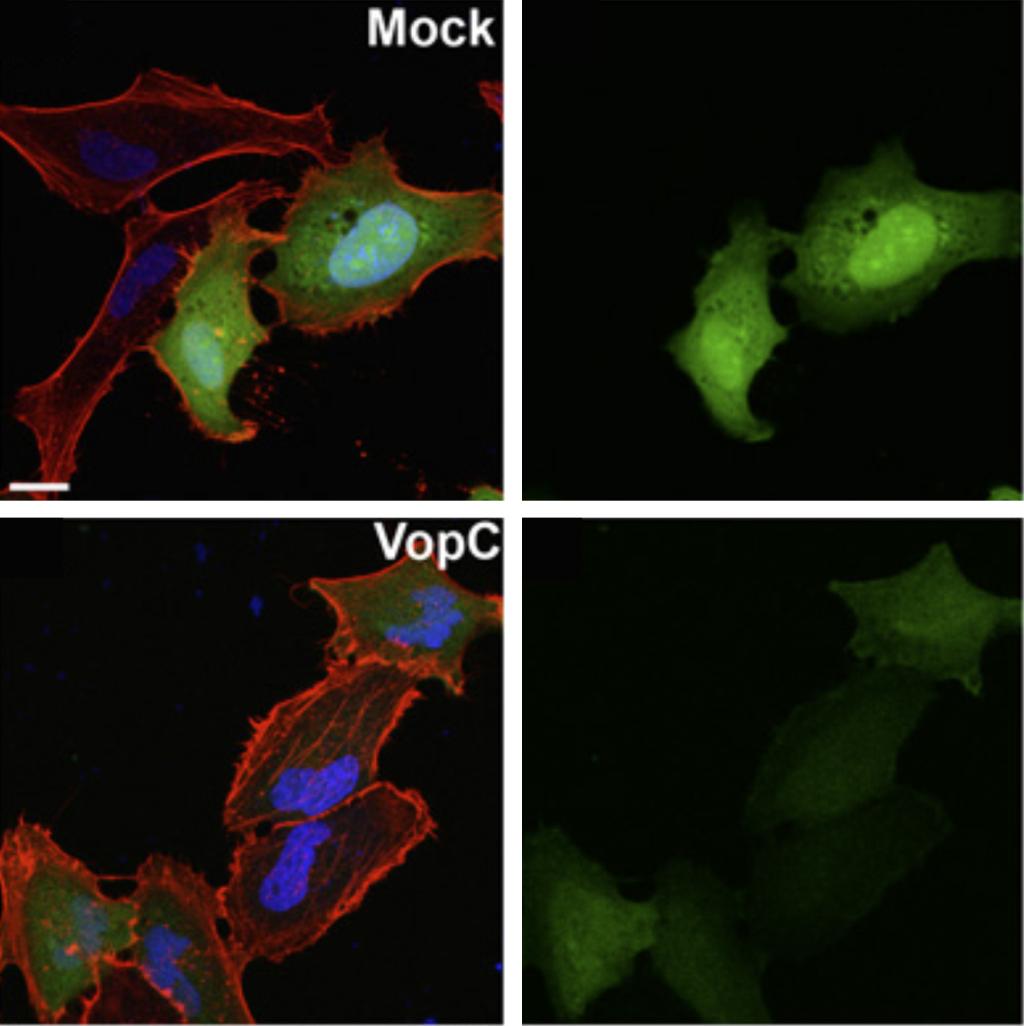
The Vpara genome contains a second pathogenicity island - encoded type III secretion system (T3SS2) that is associated with enterotoxicity. Since its discovery, this bacterium has been regarded as being exclusive extracellular. However, we demonstrated that Vpara is actually a facultative intracellular pathogen. The novel understanding of Vpara as an intracellular bacterium compares to a “rediscovery” that presents itself as a model poised for future studies of T3SS-mediated disruption of intracellular processes. Putative effectors have since been reassessed in this light and have been shown to contribute to the bacterium’s intracellular lifestyle. We have shown VopC deamidation activity on target host cell GTPases is required for this intracellular invasion. However, this activity can not be exchanged with a homologous effector from E. coli (CNF1) that also deamidates small GTPases without compromising the invasion process.
VtrA/VtrC Co-component Signaling System
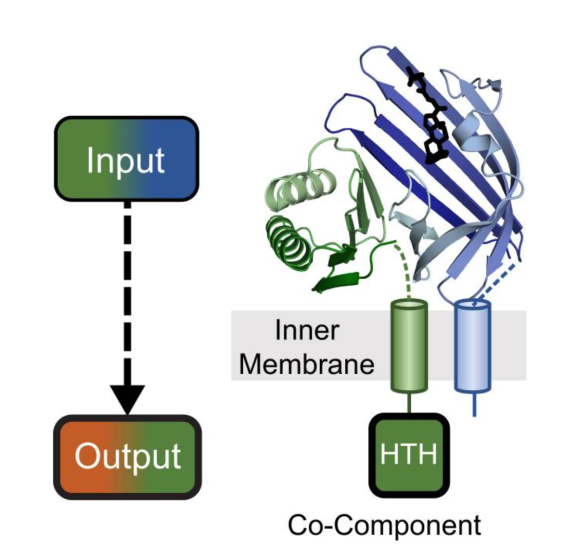
VtrA/VtrC Co-Component Signaling System
We discovered a fast-evolving co-component signaling system related to VtrA/VtrC that is conserved in many enteric bacteria. The system forms an obligate integral membrane complex of VtrC, which senses bile acids in the periplasm, and VtrA that binds target DNA in the cytoplasm to control transcription of the T3SS2 machinery and virulence effectors. Our structure studies on the VtrA/VtrC periplasmic domains have revealed the mechanism of its differential activation by TDC (activator) and CDC (antagonist), which both bind to the same site in VtrC. We are actively working on how the VtrA/VtrC periplasmic domains transduce their signal to the cytoplasmic DNA binding domain. Because this signaling system remains at the bacterial membrane, we have also studied its role in transertion, or coupled transcription, translation, and membrane insertion of target genes. A similar process may be used by co-component signaling systems from other enteric bacteria.
Orth Lab Research Highlights in Fic Domains
We discovered a bacterial virulence factor VopS from Vpara that modifies a conserved threonine residue on eukaryotic substrates via a phosphodiester bond with AMP. This modification, called AMPylation, is mediated by a domain called filamentation induced by cAMP (Fic). We have solved the structure of the VopS virulence factor Fic domain and characterized the kinetics of AMPylation as a direct transfer mechanism. As Fic domains are evolutionarily conserved in metazoans, we demonstrated this post-translational modification in eukaryotes.
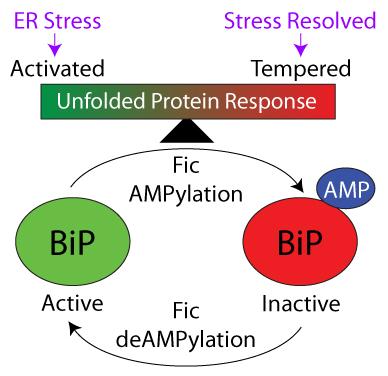
AMPylation by Eukaryotic FicD tempers the UPR
Eukaryotic FicD is localized in the endoplasmic reticulum (ER) and reversibly AMPylates BiP/GRP78 to maintain ER protein homeostasis and modulate the unfolded protein response (UPR). Prolonged or strong UPR responses can result in elevated inflammation and cellular damage. Our studies in fly demonstrated that adaptation to constant light requires Fic-mediated AMPylation of BiP to protect against reversible photoreceptor degeneration. We have also generated FicD knockout mice and evaluated the effect of FicD loss on different mouse tissues, including the exocrine pancreas and the liver. FicD tempering of the unfolded protein response is important for maintaining cellular function and avoiding tissue damage in the terminally differentiated pancreas. Liver tissue, which is more regenerative, remained remarkably resilient in the absence of FicD.
Fic-mediated AMPylation of BiP tempers the unfolded protein response
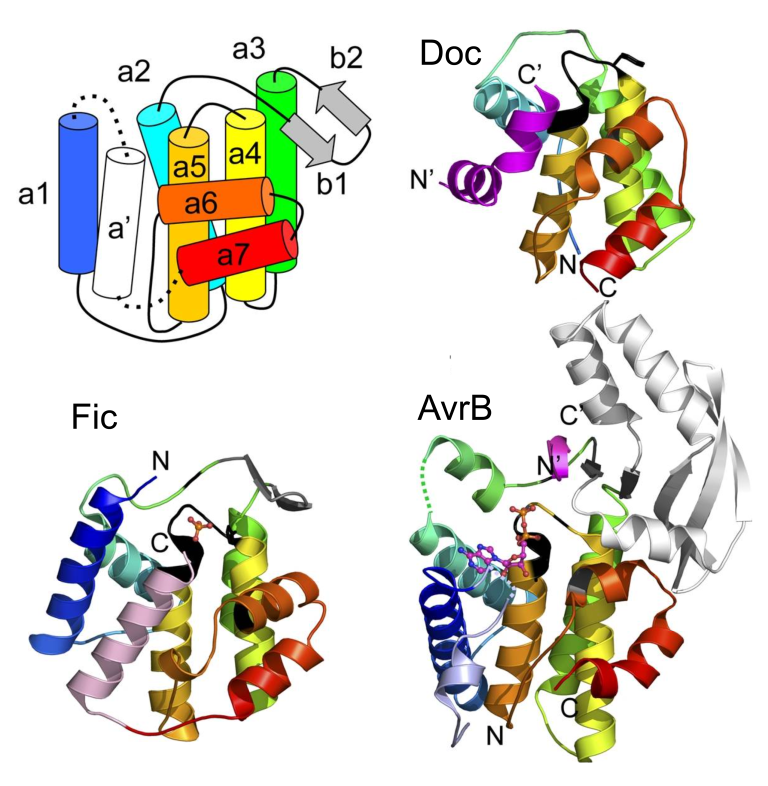
Novel Chemistry for Fic Domain Protein AvrB
Fic domains encompass a large superfamily of enzymes (including Fic Doc, and AvrB) we called FiDo for the founding members Fic and Doc. The FiDo superfamily has rapidly evolved to adopt various chemistries. We and others discovered FiDo domains can perform different post-translational modifications of protein substrates; including AMPylation, phosphocholination, UMPylation and phosphorylation. We recently uncovered the activity for one of the most elusive members of the Fic domain superfamily, AvrB. AvrB is an avirulence effector from the plant pathogen Pseudomonas syringae. Using a reliable strategy for identifying the activity of unknown effectors, we discovered AvrB uses UDP-rhamnose to modify a threonine with on its target protein RIN4 with the sugar rhamnose. Such a strategy can be applied to other rapidly evolving effectors of unknown function to unravel new activities.
FiDo superfamily conserved domain topology
Other Research
The rice blast fungus, Magnaporthe oryzae, causes one of the most destructive diseases of cultivated rice in the world. Infections caused by this recalcitrant pathogen leads to the annual destruction of approximately 10-30% of the rice harvested globally. We are interested in understanding virulence factors that contribute to host pathogenesis.
Cancer evolves through a multistep process that occurs by the temporal accumulation of genetic mutations. Tumor-derived exosomes are emerging contributors to tumorigenesis. To understand how exosomes might contribute to cell transformation, we utilized the classic two-step NIH/3T3 cell transformation assay and observed that exosomes isolated from pancreatic cancer cells, but not normal human cells, can initiate malignant cell transformation and these transformed cells formed tumors in vivo. However, cancer cell exosomes are unable to transform cells alone or to act as a promoter of cell transformation. Utilizing proteomics and exome sequencing, we discovered cancer cell exosomes act as an initiator by inducing random mutations in recipient cells. Cells from the pool of randomly mutated cells are driven to transformation by a classic promoter resulting in foci, each of which encode a unique genetic profile. Our studies describe a novel molecular understanding of how cancer cell exosomes contribute to cell transformation.
We have identified a factor involved in initial binding of host cells by a wide range of Gram-negative pathogens, Multivalent Adhesion Molecule (MAM) 7. Interference with MAM7-mediated attachment, by pre-incubation of host cells with recombinant MAM7, significantly delays the onset of hallmarks of infection. Targeting or exploiting MAM7 might provide a valuable tool for combating Gram-negative bacterial infections and we are currently exploring applications of MAM7 as an antimicrobial agent.