-
Neurodegeneration I Cell death
-
Mitochondrial dysfunction I Oxidative stress
-
PARP I DNA damage I Aging-related genome instability
-
Alzheimer's disease I Stroke I Traumatic brain injury
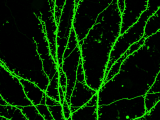
Wang Lab studies the molecular, cellular, and metabolic mechanisms of neurodegeneration and cell death using in vitro and in vivo models of neurological diseases including Alzheimer's disease, stroke, and traumatic brain injury as well as human cancers.
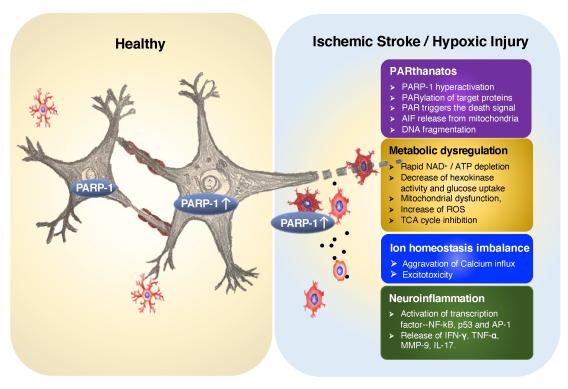
The current research topics are i) mitochondrial dysfunction and neurodegeneration in Alzheimer's disease; ii) PARthanatos (PARP-1-dependent cell death) and its post-brain injury effect on neurodegeneration and dementia in young and aged animals; iii) biological functions of PARP, apoptosis-inducing factor (AIF) and newly identified key PARthanatos regulators in health and diseases.
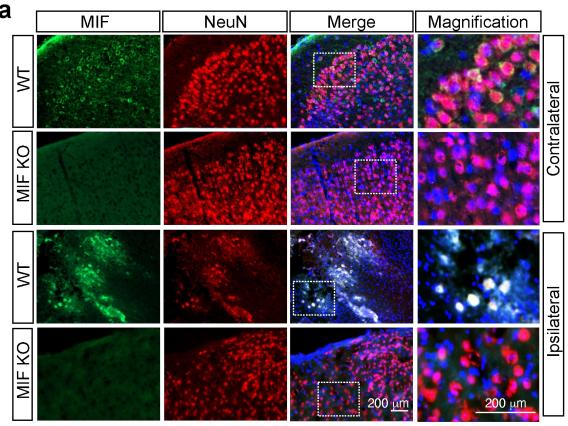
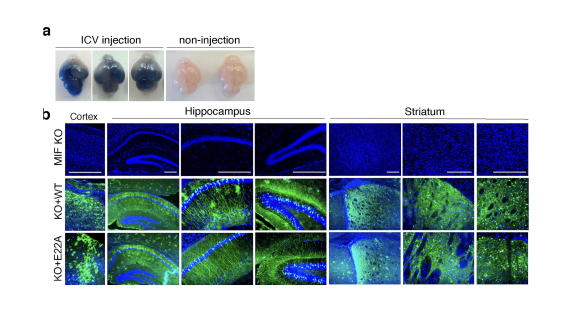
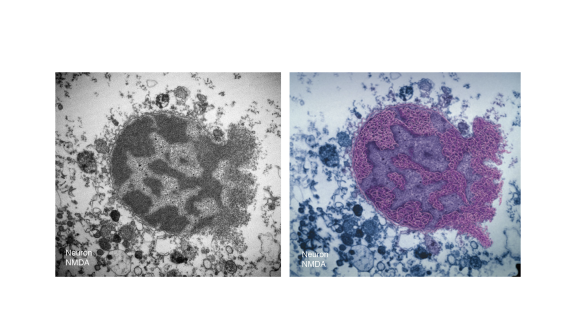
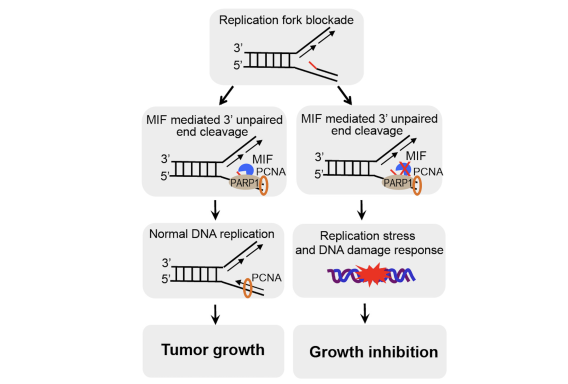
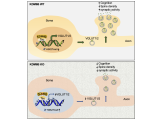
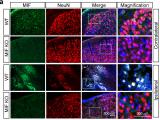
PARP-1 is a DNA damage sensor. It plays a central role in PARthanatic cell death through a caspase-independent manner after ischemia-reperfusion injury, glutamate excitotoxicity and various inflammatory responses, as well as other neurological and non-neurological diseases. Apoptosis-inducing factor (AIF) is the key mediator of PARthantos. We recently identified macrophage migration inhibitory factor (MIF) as a novel PARP-1 activity associated nuclease (PAAN) and the executor of parthanatos (Wang Y., et al. Science, 2016).
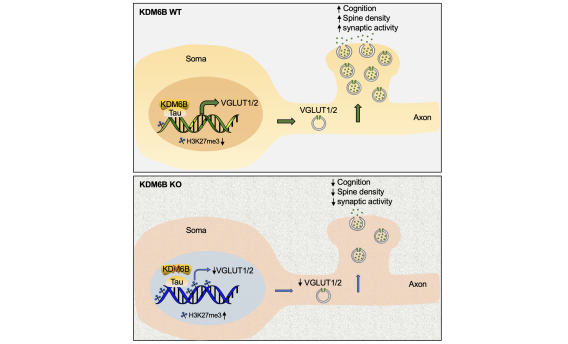
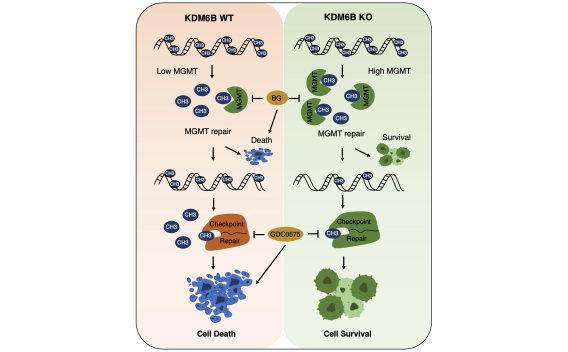
To study how PARthanatos is regulated, we performed unbiased whole genome screens using both loss-of-function and gain-of-function CRISPR technique, and identified a short list of genes positively or negatively regulating PARP-1 signaling and PARthanatos, including the epigenetic factor histone H3K27 demethylase 6B (KDM6B). We found that KDM6B regulated the excitatory neurotransmitter glutamate release in neurons via directly demethylating H3K27me3 at the promoters of vesicular glutamate transporters vGlut1 and vGlut2 to induce their expression (Wang Y., et al., Molecular Psychiatry, 2022). Currently, we are studying functions of KDM6B as well as other newly identified key regulators in PARthanatos in different types of neurological diseases including Alzheimer's disease, stroke and traumatic brain injury.
Our goals are to obtain a comprehensive molecular understanding of PARthanatos signaling regulation in neurological diseases and to prevent/delay neuronal cell death.
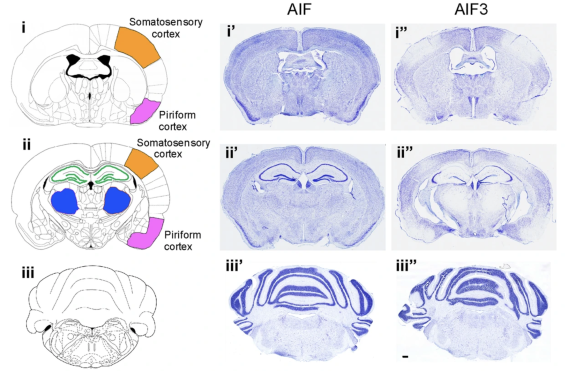
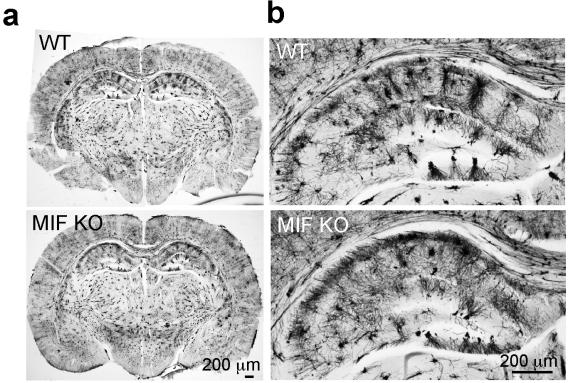
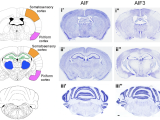
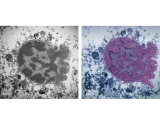
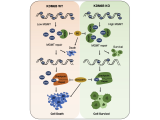
Apoptosis-inducing factor (AIF) is an X-chromosome linked mitochondrial flavoprotein serving as a free radical scavenger and plays a vital function in mitochondrial bioenergetics. Previous works from the PI have revealed that AIF release from the mitochondria and translocation to the nucleus mediates PARP-1 dependent cell death (parthanatos) following ischemic brain injury. Spontaneous genetic mutations of AIF have been observed in both human and mouse and provided strong association of Aif gene with the mitochondrial dysfunction and etiology of neuropathy. Recently, we discovered a hitherto unknown AIF splicing isoform, defined as AIF3 distinct to other two known isoforms. Induction of AIF3 causes mitochondrial dysfunction and neuron loss in cortex and hippocampus. Our goals are to 1) obtain a comprehensive understanding of AIF3 functions in mitochondrial bioenergetics; 2) dissect the molecular and cellular mechanisms of AIF3 mediated mitochondrial dysfunction; 3) understand the role of AIF3-meidated mitochondrial dysfunction on neuronal cell death.
Alzheimer's disease (AD) is a leading cause of dementia, which is characterized by memory loss and thinking problems interfering with daily life. Although the importance of beta amyloid and Tau in AD pathogenesis has been well appreciated, unfortunately no effective treatment is available to prevent dementia progress so far due to the lack of understanding of neurodegeneration in AD. Our Lab has identified AIF3 as a potential new risk factor for neurodegeneration and will study its role and underlying molecular mechanisms of neurodegeneration in AD, with the hope to develop new therapeutic approaches to prevent or delay disease progress.
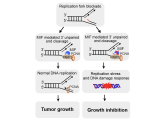
PARP-1 plays important roles in ischemic brain injury as well as cancers. Both types of diseases involve unique microenvironment-ischemia or hypoxia, which might significantly alter PARP-1 functions and signaling. In collaboration with Dr. Weibo Luo Lab, we will study how PARP-1 signaling is differential regulated under normoxia and hypoxia/ischemia conditions using ischemic stroke models as well as hypoxic-cancer models.




Join Our Lab
Our lab is currently recruiting self motivated postdoctoral fellows and graduate students. If interested, please email to Dr. Wang.