Trafficking into cilia in polycystic kidney disease (PKD)
The primary cilium has been found to be associated with a number of cellular signaling pathways, such as vertebrate hedgehog signaling, and implicated in the pathogenesis of diseases affecting multiple organs including the neural tube, kidney, and brain. The primary cilium is the site where a subset of the cell's membrane proteins is enriched. Processes determining the level of proteins in the ciliary membrane include entry into the compartment, removal, and retention by diffusion barriers such as the transition zone. However, pathways that target and concentrate membrane proteins in cilia are not well understood.
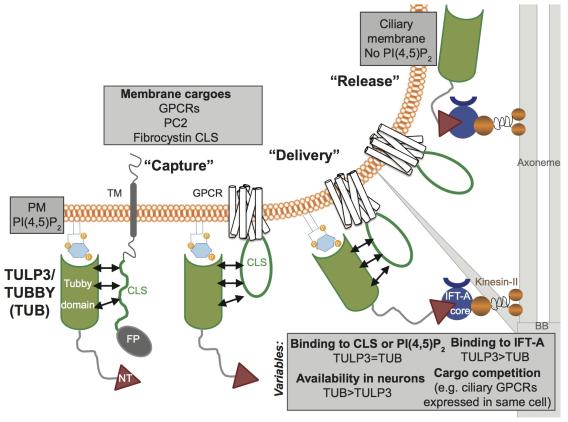 Figure 1. Model for TULP3-mediated trafficking to cilia. Abbreviations: BB, basal body; CLS, ciliary localization signal; FP, fusion protein; NT, TULP3 IFT-A binding N-terminus domain.
Figure 1. Model for TULP3-mediated trafficking to cilia. Abbreviations: BB, basal body; CLS, ciliary localization signal; FP, fusion protein; NT, TULP3 IFT-A binding N-terminus domain. The tubby family proteins, Tulp3 and tubby (Tub) have been implicated as adapters in trafficking of multiple GPCRs into the ciliary membrane (Loktev and Jackson, 2013; Mukhopadhyay et al., 2013; Sun et al., 2012). These tubby family proteins have a N-terminal intraflagellar complex A (IFT-A) core binding conserved helix, and a C-terminal tubby domain that binds to PI(4,5)P2 (Mukhopadhyay et al., 2010; Santagata et al., 2001). Disrupting either of these domains prevents trafficking of these GPCRs to cilia, suggesting that Tulp3 “bridges” the GPCRs with IFT-A core in targeting them into cilia (Mukhopadhyay et al., 2010). We have recently shown that TULP3 to function as a general adapter for ciliary trafficking of structurally diverse integral membrane cargo, including multiple reported and novel rhodopsin family GPCRs, and polycystic kidney disease causing polycystin-1/2 complex. TUB also localizes to cilia similar to TULP3, and determines trafficking of a subset of these GPCRs to neuronal cilia. Using minimal ciliary localization sequences from GPCRs, and fibrocystin, also implicated in polycystic kidney disease, we demonstrate these motifs to be sufficient and TULP3-dependent for ciliary trafficking. We propose a three-step model for TULP3/TUB-mediated ciliary trafficking, including capture of diverse membrane cargo by tubby domain in a phosphoinositide 4,5-bisphosphate (PI(4,5)P2)-dependent manner, ciliary delivery by intraflagellar transport complex-A binding to TULP3/TUB N-terminus, and subsequent release into PI(4,5)P2-deficient ciliary membrane (Badgandi et al., 2017; Mukhopadhyay et al., 2010; Mukhopadhyay et al., 2013). Understanding the trafficking mechanisms that underlie the role of ciliary compartmentalization in signaling might provide unique approaches for intervention in progressive ciliopathies.
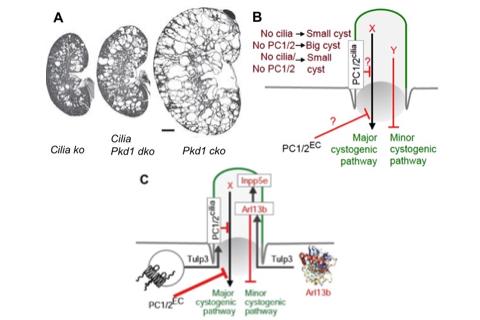 Figure. 2. A-B. Cystogenesis from PC1/2 loss is more severe and is suppressed from cilia loss. C. Cystogenesis upon Tulp3 deletion was intermediate between that caused by loss of PC-1 or cilia (Figure 3). Defects in Arl13b trafficking to cilia precede cystogenesis from Tulp3 loss (Figure 4). Arl13b might be the cilia-localized negative regulator of cystogenesis functioning independent of PC1/2. Kidney images from (Ma et al., 2017). EC, extraciliary; dko, double knockout.
Figure. 2. A-B. Cystogenesis from PC1/2 loss is more severe and is suppressed from cilia loss. C. Cystogenesis upon Tulp3 deletion was intermediate between that caused by loss of PC-1 or cilia (Figure 3). Defects in Arl13b trafficking to cilia precede cystogenesis from Tulp3 loss (Figure 4). Arl13b might be the cilia-localized negative regulator of cystogenesis functioning independent of PC1/2. Kidney images from (Ma et al., 2017). EC, extraciliary; dko, double knockout. Ciliary trafficking in polycystic kidney disease
Polycystic kidney disease proteins, polycystin-1 and polycystin-2, localize to primary cilia. Polycystin knockouts have severe cystogenesis compared to ciliary disruption, whereas simultaneous ciliary loss suppresses excessive cyst growth. These data suggest the presence of a cystogenic activator that is inhibited by polycystins, and an independent but relatively minor cystogenic inhibitor, both of which are cilia-dependent (Ma et al., 2017) (Figure 2). However, current genetic models targeting cilia completely ablate the compartment making it difficult to uncouple cystoprotein function from ciliary localization. Thus, the role of cilium-generated signaling in cystogenesis is unclear.
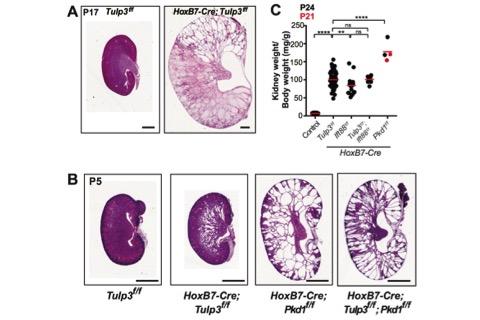 Figure 3. Tulp3 cko cysts are smaller than Pkd1 cko cysts.
Figure 3. Tulp3 cko cysts are smaller than Pkd1 cko cysts. Cystogenesis in Tulp3 cko.
As polycystins and fibrocystin CLS fusions are affected upon Tulp3 loss, we hypothesized that loss of Tulp3 would result in renal cystogenesis. We found that of kidney-specific Tulp3 conditional
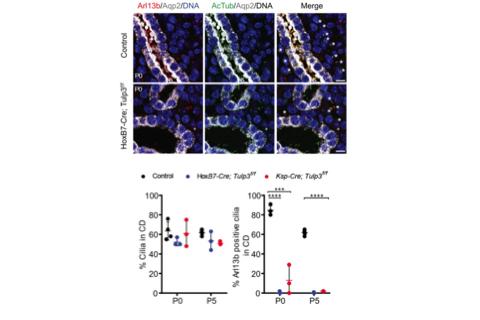 Figure 4. Loss of Arl13b in cilia in Tulp3 cko before cystogenesis, P0 kidneys, left.
Figure 4. Loss of Arl13b in cilia in Tulp3 cko before cystogenesis, P0 kidneys, left. knockout (cko) using two mouse models (Ksp1-Cre; Tulp3f/f (Ksp-Tulp3 cko) and HoxB7-Cre; Tulp3f/f (HoxB7-Tulp3 cko)) resulted in cystogenesis (Figure 3A). Based on kidney to body weight ratio, cystic index and epithelial proliferation in developing tubules/cysts the severity of cystogenesis was intermediate between that caused by loss of polycystin-1 or cilia (Figure 3B-C). The Somlo lab demonstrated that inhibition of cilia formation in Pkd1/2 cko rescued cyst formation (Ma et al., 2013) (Figure 2A). However, concomitant Tulp3 loss did not inhibit cystogenesis in Pkd1 cko, unlike ciliary disruption (Figure 3C). Thus, at least some of Tulp3 functions in kidney are independent of its role in PC1 ciliary transport (Hwang et al., 2019). Defective Arl13b trafficking to intact cilia precedes cyst formation in Tulp3 cko kidney collecting ducts. Cilia from epithelial cells lining the cysts in Tulp3 cko are not affected during cyst initiation or at later stages. We thus hypothesized that defective ciliary trafficking of proteins in addition to PC2 might account for the intermediate cystic disease phenotypes. One candidate is the small GTPase Arl13b, because of its role in causing cystogenesis, similar in timeline and extent of cystogenesis compared to loss of Tulp3. Trafficking of Arl13b to cilia was dramatically reduced in either Hoxb7-Tulp3 or Ksp-Tulp3 cko collecting duct epithelia. Interestingly, this defect was evident as early as P0, well before cysts were apparent in either model (Figure 4). Adjacent Aqp2-negative nephron segments still retained Arl13b-positive cilia at these stages (Figure 4, asterisks), confirming that the trafficking defect is primarily in collecting ducts. We propose that the kidney cystogenesis in Tulp3 cko models resulted from cumulative loss of PC2 and Arl13b in cilia. Furthermore, Tulp3 is the major factor regulating trafficking of Arl13b into kidney collecting duct cilia (Hwang et al., 2019). With the discovery of Tulp3 as a potential gatekeeper of the ciliary lipidated proteome, we are uniquely positioned to determine the role of ciliary trafficking of diverse Tulp3-regulated cargoes in the multiple developmental processes and disease conditions.
References
Badgandi, H.B., Hwang, S.H., Shimada, I.S., Loriot, E., and Mukhopadhyay, S. (2017). Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J Cell Biol 216, 743-760.
Hwang, S.H., Somatilaka, B.N., Badgandi, H., Palicharla, V., Walker, R., Shelton, J.N., Qian, F., and Mukhopadhyay, S. (2019). Tulp3 regulates renal cystogenesis by trafficking of cystoproteins to cilia. Curr Biol In Press.
Loktev, A.V., and Jackson, P.K. (2013). Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep 5, 1316-1329.
Ma, M., Gallagher, A.R., and Somlo, S. (2017). Ciliary Mechanisms of Cyst Formation in Polycystic Kidney Disease. Cold Spring Harb Perspect Biol.
Ma, M., Tian, X., Igarashi, P., Pazour, G.J., and Somlo, S. (2013). Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45, 1004-1012.
Mukhopadhyay, S., Wen, X., Chih, B., Nelson, C.D., Lane, W.S., Scales, S.J., and Jackson, P.K. (2010). TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev 24, 2180-2193.
Mukhopadhyay, S., Wen, X., Ratti, N., Loktev, A., Rangell, L., Scales, S.J., and Jackson, P.K. (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210-223.
Santagata, S., Boggon, T.J., Baird, C.L., Gomez, C.A., Zhao, J., Shan, W.S., Myszka, D.G., and Shapiro, L. (2001). G-protein signaling through tubby proteins. Science 292, 2041-2050.
Sun, X., Haley, J., Bulgakov, O.V., Cai, X., McGinnis, J., and Li, T. (2012). Tubby is required for trafficking G protein-coupled receptors to neuronal cilia. Cilia 1, 21.